

1. Introduction
Congenital chloride diarrhea is an autosomal recessive disorder caused by mutations in SLC26A3 gene encoding intestinal epithelial Cl-/HCO3- exchanger that is coupled with Na+/H+ exchanger. These defects result in excessive chloride rich diarrhea, dehydration and hypochloremic, hypokalemic metabolic alkalosis. Congenital chloride diarrhea was defined in 1945 and around 260 cases have been reported in literature until today mostly from Finland, Poland and Arabic peninsula. Polyhydramnios, lack of meconium, weight loss and failure to thrive are other characteristics of the condition. Diagnosis is based on clinical features with fecal chloride concentration > 90 mmol/L. Other important causes to be excluded in neonatal and infant period are those in which hypoelectrolytemia is due to loss through urine in Bartter syndrome, sweat in cystic fibrosis and vomiting in pyloric stenosis. Early diagnosis of congenital chloride diarrhea with differentiation from other diseases is crucial because life-long fluid, sodium and potassium replacement is required to avoid renal damage and morbidity (1, 2).
2. Case Presentation
A 13-month-old female patient presented with complaints of vomiting, diarrhea, and growth failure. She experienced vomiting 1 - 2 times a day and watery stools 10 - 15 times a day, which did not contain mucus or blood, for 4 days. Physical examination revealed weakness, malnutrition (weight: < 3rd percentile, -3.35 SDS; height: < 3rd percentile, -2.83 SDS; head circumference: < 3rd percentile, -1.61 SDS), and blood pressure was 85/46 mm Hg.
She was diagnosed with Bartter syndrome at another center when she was 3 months old and had been using KCl tablets, hypertonic NaCl, and indomethacin for 10 months. She was born at 36 weeks of gestation, weighing 2590 g, with a history of polyhydramnios. There was second-degree consanguinity between the parents, with no similar problems in relatives.
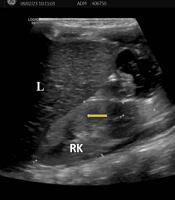
Laboratory investigations were as follows: Complete blood count was normal; routine urine analysis showed pH 9, density 1005, protein 1+, microscopy 4 - 6 leukocytes/area; serum urea was 89 mg/dL, creatinine 0.65 mg/dL, uric acid 13.4 mg/dL (normal: 2.6 - 6), sodium 136 mmol/L, potassium 2.4 mmol/L, chloride 80 mmol/L; blood gas analysis showed pH 7.65, bicarbonate 37 mmol/L, base excess +13 mmol/L; spot urine protein/creatinine ratio was 0.6 mg/mg. Rotavirus antigen was positive in stool, while stool and urine cultures were negative. She had hypokalemic hypochloremic metabolic alkalosis and proteinuria. Renal ultrasonography revealed increased echogenicity of the kidneys with loss of corticomedullary differentiation (Figure 1).
; (Abbreviations: L, liver; RK, right kidney).")
Renal ultrasonography revealing increased kidney echogenicity with loss of corticomedullary differentiation (arrow); (Abbreviations: L, liver; RK, right kidney).
We continued her medications and added intravenous fluid replacement for Bartter syndrome with rotavirus diarrhea. However, hypochloremic metabolic alkalosis and diarrhea continued 6 - 7 times a day, which could not be corrected after the initiation of indomethacin. The history of polyhydramnios and diarrhea since birth without polyuria indicated that the diagnosis of Bartter syndrome should be reconsidered. Fecal electrolytes and genetic analysis were planned. We could not obtain urine electrolyte results from the previous medical center, but the results we obtained were: Urine sodium 46 mmol/L, potassium 84 mmol/L, chloride 14 mmol/L (low < 20), calcium/creatinine ratio 0.14 mg/mg (normal < 0.53). Serum renin was 136 ng/mL/s (normal: 0.51 - 2.64), aldosterone 2710 pg/mL (normal: 50 - 900). Fecal electrolytes were: Sodium 86 mmol/L (normal: 20 - 40), potassium 54 mmol/L (normal: 55 - 65), chloride 154 mmol/L (normal: 10 - 20, > 90 diagnostic for congenital chloride diarrhea). Urine chloride was low despite low serum chloride levels. Fecal sodium and chloride levels were high. Hearing test was normal, and nephrocalcinosis was not detected. A sweat chloride test could not be performed; however, immunoreactive trypsin was normal in the neonatal screening test to rule out cystic fibrosis, and genetic analysis was available. Whole exome sequencing revealed a nonsense homozygous SLC26A3 c.559G>T p.(Gly187*) (rs121913032) mutation compatible with autosomal recessive congenital chloride diarrhea and classified as a pathogenic variant. Familial segregation analyses by Sanger sequencing confirmed the heterozygous states of the parents. Genetic counseling was provided, informing the parents that amniocentesis would be required if they plan a second pregnancy.
3. Discussion
Bartter syndrome diagnosis should be elucidated in hypoelectrolytemic metabolic alkalosis to differentiate it from pseudobartter syndrome. Specifically, cystic fibrosis and congenital chloride diarrhea should be considered for differential diagnosis (2). Urine chloride level and response to chloride replacement are of major importance. Urine and stool are generally mixed in infancy. Recurrent episodes of diarrhea should be questioned in detail repeatedly. Sometimes watery stool may be mistaken for urine output by parents and not noticed for some period, as in the presented case, leading to a delay in diagnosis. Urine obtained to measure electrolyte levels reveals high values because samples contain stool. Prolonged dehydration may lead to glomerulosclerosis, hyalinosis, proteinuria, and renal function loss (3). The presence of diarrhea with stool and urine electrolyte investigation should not be missed. Our patient had a low urine chloride level despite hypochloremia. The most valuable electrolyte evaluation in this condition is urine chloride. Urine sodium and potassium levels may be high even if their serum levels are low. Excretion of bicarbonate due to metabolic alkalosis increases urinary loss of sodium. This induces aldosterone synthesis and increases potassium loss (2, 4). Our case had a high urine potassium level, and urine pH was 9. If sodium delivery is low in the distal nephron, urine may become alkaline due to inhibition of the H+ pump from renal A intercalated cells. Also, high urine pH may reduce metabolic alkalosis in congenital chloride diarrhea by modulation of renal Cl-/HCO3- exchange (5).
Common features of Bartter syndrome and congenital chloride diarrhea include polyhydramnios, growth retardation, malnutrition, failure to thrive, hypoelectrolytemia, and metabolic alkalosis. However, ongoing diarrhea, hypoelectrolytemic metabolic alkalosis, and low urine chloride despite Bartter syndrome treatment with indomethacin in the current case led to reinvestigation of the diagnosis by checking fecal electrolytes and genetic analysis. To date, more than 30 mutations of the SLC26A3 gene compatible with congenital chloride diarrhea have been reported without a phenotype-genotype correlation (5, 6).
Interestingly, an infant case was shown to have cystic fibrosis and congenital chloride diarrhea simultaneously. This baby had ongoing hypoelectrolytemia and metabolic alkalosis after being followed up and treated for cystic fibrosis. Further investigation showed low urine electrolytes, hyperreninemia, and hyperaldosteronemia. Endoscopy and fecal electrolytes were checked due to the persistence of diarrhea, and an additional diagnosis of congenital chloride diarrhea was made (2).
Another rare case had both type 4 Bartter syndrome and congenital chloride diarrhea, presenting with hypokalemia, hypochloremic metabolic alkalosis, hyponatremia, polyuria, hearing loss, and nephrocalcinosis, as expected in type 4 Bartter syndrome. Urine chloride, potassium, and calcium were increased. Indomethacin and spironolactone were initiated. Daily diarrhea continued 12 - 15 times, and fecal chloride concentration was found to be high (152 mmol/L). Despite the lack of genetic confirmation, this 8-month-old boy was diagnosed with type 4 Bartter syndrome and congenital chloride diarrhea (7). While the majority of congenital chloride diarrhea cases have been detected in infancy, a few have been diagnosed during adolescence. A 28-year-old man was misdiagnosed with Bartter syndrome since 5 months of age based on dehydration, hypokalemia, metabolic alkalosis, and a sister who was also misdiagnosed with Bartter syndrome. He had excessive diarrhea, abdominal pain, intestinal ulcers, and perianal fistula and was diagnosed with ulcerative colitis during adolescence. He developed chronic renal failure at 18 years due to recurrent episodes of dehydration and had a kidney transplantation at 21 years of age. Persistent metabolic alkalosis with diarrhea after transplantation was unusual, and genetic testing detected congenital chloride diarrhea. Overlap of ulcerative colitis with congenital chloride diarrhea and a misleading family history of Bartter syndrome were factors leading to a delay in diagnosis (8).
Prolonged dehydration attacks, hypovolemia with activation of the renin-angiotensin-aldosterone system, lead to interstitial fibrosis and glomerular sclerosis, as described in secondary focal segmental glomerulosclerosis cases. Proteinuria and renal transplantation are reported in a few congenital chloride diarrhea and salt-losing tubulopathy patients (4, 5). Although various treatment options such as butyrate and cholestyramine have been reported but shown ineffective in reducing diarrhea, NaCl, KCl, and fluid replacement without any other dietary modification is still considered the current appropriate treatment for congenital chloride diarrhea. Butyrate is a short-chain fatty acid exerting an absorptive stimulus on intestinal NaCl transport and an anti-secretory effect on chloride secretion. The therapeutic efficacy of oral butyrate was demonstrated in seven pediatric congenital chloride diarrhea cases with different SLC26A3 genotypes. Fecal chloride loss and stool pattern were not changed with butyrate use in those with the c.559G>T mutation (9-11).
Increased renal echogenicity and loss of corticomedullary differentiation in the current case are also findings probably due to chronic hypovolemia. These findings may also be expected in acute or chronic renal failure and inflammation, as well as increased echogenicity may be a normal variant in underweight children. As proteinuria in our patient is not very high, we planned to observe and perform a renal biopsy and initiate angiotensin-converting enzyme inhibitors that have an antiproteinuric effect if proteinuria increases. Hypovolemia causes activation of the renin-angiotensin-aldosterone system, leading to glomerular hyperfiltration, so proteinuria occurs secondary to hypovolemia. We suggest that follow-up for proteinuria is important, and a decrease in proteinuria depends on fluid replacement. The aim and success of appropriate treatment with fluid and electrolyte replacement is the normalization of serum electrolytes, metabolic alkalosis, and proteinuria. Our patient is on sufficient water and 4 - 5 meq/kg/day KCl and 2 - 3 meq/kg/day NaCl replacement. Indomethacin was discontinued. Her growth and development have improved after 5 months (weight: < 3rd percentile, -2.32 SDS; height: 3rd - 10th percentile, -1.8 SDS; head circumference: 10th percentile, -1.32 SDS). Renal function tests normalized with episodes of rehydration. The long-term management plan should focus on monitoring renal functions, which may determine the overall outcome of the disease.
3.1. Conclusions
In conclusion, congenital chloride diarrhea may be misdiagnosed as Bartter syndrome in infancy. Fluid-electrolyte loss, dehydration, and renal ultrasound findings are common features of congenital chloride diarrhea and Bartter syndrome, making diagnosis challenging. Watery stool may be mistaken for urine by parents of congenital chloride diarrhea cases. A low urine chloride level despite hypochloremia is the most valuable test for differential diagnosis. As chronic hypovolemia is a cause of proteinuria and renal damage, diagnosis is essential to prevent significant morbidity and mortality. We aimed to draw attention not only to Bartter and pseudo-Bartter syndrome differentiation but also to the need for further care on proteinuria, hence the progression of renal damage.
