

1. Definitions
Intellectual disability (ID) is a general term for mental retardation (MR) with significant impairment of cognitive and adaptive development (1). It is a lifelong disability that affects about 1% to 3% of the universal population. It imposes a heavy burden on affected families, the society, and the health care system (2, 3). Extensive evidence indicated that in particular regions, by consanguinity, recessive mutations have high incidence rates (4). Autosomal dominant (AD) de novo mutations occur in sporadic forms of ID cases. A consanguineous family study showed the detection of pathogenic variants in many autosomal recessive (AR) disorders (5). According to large-scale autozygosity mapping, intellectual disability in AR type has an extremely heterogeneous molecular basis; some evidences suggests that approximately 3000 human genes are related to autosomal recessive ID (6, 7). Today, the application of ID terminology has changed. Also, ‘‘developmental delay’’ refers to a disorder in children and in only one or more developmental function, like several delays in the prevalent acquisition activity, which is affected by the natural development (4). This term refers to the limitation, condition in trustworthy, cognition and validity.
Due to the genetic diversity of multiple disorders, genomes molecular evaluation is required for the entire world (8-10). While diagnostic methods are developed, yet the wide majority of ID causes do not have definitive molecular detection.
2. Intellectual Disability Classification
The evaluation of the child with a developmental disorder plays a key role in early ID diagnosis. Furthermore, ID is determined as follows: First, through examination of IQ (intelligence quotient), defined by a total human intelligence score derived from standardized tests (below 70); second, according to developmental symptoms, including cognition, motor performance, hearing, speech and vision that begin before 18 years old; third, according to limitations in environment adaptation, including self-care and skills of communication (11). Intellectual disability is divided to four subgroups, according to IQ test: (I) The mild group (IQ: 50 to 70), (II) the moderate group (IQ: 35 to 49), (III) the severe group (IQ: 20 to 34), and (IV) the profound group (IQ below 20) (12, 13). Most of the patients belong to the mild group with an 85% incidence rate, the moderate group as the second prevalent ID has an incidence rate of 10%, and only 5% of ID are included in the severe and profound group (14). Terminal ID can be presented to syndromes (S) and non-syndromes (NS). The NS patients have sole clinical abnormalities without any associated features, therefore, S patients show further dysmorphological features, such as abnormalities of brain structure or function, metabolic-pathway defect, and skeletal anomalies (15).
3. Etiology
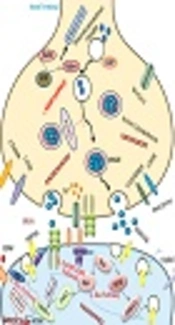
The perspective of disease etiology can provide beneficial and critical clinical data for diagnostic, prognostic, concept, and convert to treatment. This concept helps couples in planning and explaining the recurrence risk of the next pregnancy in the near future. This study briefly explains several causes of intellectual disability and global developmental delay. Two main ID etiologic factors are genetic factors and non-genetic factors (Figure 1) (16, 17).
 and non-genetic factors (I, prenatal disorders; II, environmental factors; III, infections; IV, childhood illness)")
Two main of ID etiologic genetic factors (I, chromosome aberrations; II, fragile X syndrome; III, defect of imprinted genes; IV, de-novo mutations) and non-genetic factors (I, prenatal disorders; II, environmental factors; III, infections; IV, childhood illness)
3.1. Hereditary Factors
3.1.1. Chromosomal Abnormalities
Chromosomal abnormalities are represented by various percentages in different regions of the world that could be diagnosed with conventional cytogenetic procure (16). If the range of XLMR in moderate to severe form is lower than 0.1%, this means that the account of X-linked ID is 10% to 16% of the all severe patients (18). Down syndrome is the major anomaly involving chromosome trisomy 21 with an incidence rate of one to 700 (19). Edwards syndromes (trisomy 18) has a 1 to 4000 prevalence (20), and this trisomy typically leads to death in the first days of birth. In addition, all monosomies in the autosomal chromosome lead to miscarriage in the primary embryonic stage yet microdeletion, such as cri-du-chat syndrome (5p microdeletion), survive to adulthood with significant disability and Wolf-Hirschhorn syndrome (4p microdeletion) survives until their 20s with sever disability (21). Contiguous gene syndrome (CGS) is explained by chromosome abnormalities in the chromosomes, such as deletion and duplications, and leads to alteration of normal gene dosage. Some CGS include Miller-Dieker syndrome (17p13.3 deletion), Williams’s syndrome (7q11.23q11.23 deletion), Di George syndrome (22q11 deletion), and Langer-Giedion syndrome (8q24.1 deletion) (22).
3.1.2. Trinucleotide Repeats Expansion
The fragile X syndrome (FXS) is the most widespread form of syndromic X linked to mental retardation (XLMR) with an approximate occurrence of 1:5000 in males. The FXS patients have a folate-sensitive fragile site in X (q27.3), which is linked with an increase in the number of CGG trinucleotide repeats. The 5’ untranslated region (UTR) section of X (q27.3) can encode FMR1 as RNA binding protein (23). The FMR1 genes are classified to four subgroups, according to the number of CGG expansions: (I) Normal phase, including 6 to 24, (II) intermediate 25 to 54, (III) permutation 55 to 200, and (IV) fully mutated up to 200 repeats. In cases, who have more than 200 CGG triplet repeats, a down-regulation of FMR1 gene is observed (24).
3.1.3. Defects on Imprinted Genes
Genomic imprinting is an epigenetic process that causes genes to express from only single alleles inherited from each parent. This occurrence depends on DNA epigenetic modifications, such as acetylation and methylation (25). The paternal chromosome deletion in 15q11.2q13 segment can lead to Prader-Willi syndrome and maternal deletion of coding region in UBE3A (15q11.2q13) gene leads to the Angelman syndrome (26). A similar mutation cannot be the reason for MR until inherited from the father.
3.1.4. De Novo Mutations
Recent reports demonstrated that several de novo mutations (DNMs) in the brain can involve considerable pathogenic genes in the neurological disturbance. Furthermore, DNMs could be from a different origin from both parent’s chromosomes, which means genetic alternation is not transmitted from the maternal or paternal gene. Furthermore, these mutations may be recessive and dominant hereditary patterns. Some DMNs are reported in Iran by whole exome sequencing (27).
3.2. Non-Genetic Factors
3.2.1. Environmental Agents
Environmental factors, as the main particular etiology of ID, are well-known in neurobehavioral disturbance (17). Environment agents, such as nutritional deficiencies as well as alcohol and drugs abuse, cause about 5% to 20% of ID (28). Avoiding children from exposure to toxicants can prevent several developmental delays. Particular lack of nutrition may be the initial cause of ID as exemplified by the correlation of myelomeningocele and folate deficiency (29). In several cases, metabolic disorders are the probable cause of ID, including phenylketonuria and hypothyroidism, which can be prevented with primary therapy. While sphingolipidoses and mucopolysaccharidosis show less response for the initial intervention (30).
3.2.2. Birth Illness
The perinatal period begins at the end of the 22nd week of gestation and terminates within seven days after delivery. Perinatal and maternal health is closely linked together. Several complications of pregnancy comprise of mother’s diabetes, kidney disorder, and heart disease and are a possible source of ID in newborns. In addition, severe prematurity, birth asphyxia, jaundice, and birth trauma during this period may be due to mental disorder (31). Neurotoxicants influence vital developmental stages of central nervous system, including myelinization, cell proliferation, nerve cell connections, cell migration, and apoptosis (32).
3.2.3. Infections
One of the vital reasons of infant morbidity and mortality is maternal infection exposure in the gestation process that leads to neurological disruption (33). The fetus is comforted by an infection in the genital tract during delivery. The cytomegalic virus, toxoplasmosis, rubella, and herpes zoster are examples of transmission, the infection that is harmful to the fetus. The potential of infections is associated with the period of exposure during a pregnancy (34). Postnatal problems consist of the childhood and infancy period. The major causes of several brain infections are meningitis, Japanese encephalitis, and tuberculosis. Also, chronic lead exposure, head trauma and malnutrition are involved in postnatal injuries (12).
4. ID Diagnosis
This definitive assessment often needs numerous health specialists, who prepare supplementary skills to ensure a detailed evaluation. Physician collaboration with another professionalism can usually promote the validation of clinical judgment (35). The numerous causes of intellectual disability and global developmental delay are a broad challenge for practitioners. For this purpose, comprehensive guidelines have been developed for various professionals, such as medical genetics (36), the American Academy of Pediatrics, and the American Academy of Child Neurology. This guideline is performed according to existing published reports (37). Figure 2 is the suggested guideline for ID diagnosis.
 and whole exome sequencing.")
Diagnostic flow chart for evaluation of intellectual disability patients first: Family history; second: Physical examination; third: Laboratory diagnostic tests, including: Cytogenetic study, assessment of fragile X syndrome, array studies (SNP, CGH) and whole exome sequencing.
4.1. History
The child evaluation of mental retardation or developmental delay begins with a precise familial history. Genetic counselors must evaluate at least three generations and apply open-ended questions, such as prior familial infantile or neonatal loss or maternal pregnancy death. In addition, the geographic area or some ethnicities can be clues to potential causes of ID patients (38). More data is required regarding the mother’s pregnancy period, such as harmful antenatal occurrences, vaginal bleeding, adverse infections, gestational diabetes or other clinical conditions. Tobacco or alcohol use may have possible implications for fetal development (35). Vital parameters are detected such as the child’s birth appearance, activity, weight, grimace, pulse, and respiration scores; moreover, a neurologic examination is helpful for early diagnosis of the newborn. Finding this information can prepare a critical clue to the source of the child’s developmental disorders. Evaluation of the child's developmental performance in all scopes should be done in some daily activities, such as feeding, dressing, toileting, and self-hygiene (39).
4.2. Physical Examination
The physical examination is the next phase of child assessment for ID or MR. This examination often contains a major formal content and minor formal evaluation. The general and neurologic examination, consists of height, weight, and BMI measurement, and is the key formal content. Then, detailed assessments for dysmorphic appearance, which requires to be matched by ethnic and geographic conversion, is performed. The abdomen examination for possible liver or spleen enlargement is necessary. In addition, the spine should be examined for any abnormalities. Another fundamental parameter is the head circumference, which depends on gender and age. Neurologic examination includes cranial nerves, such as papillary abnormalities, facial paresis, and dysarthria and ascertaining. The motor neurologic examination evaluates possible lateralizing parameters or muscle strength, and bulk and plantar responses. Through examination, the modality of limb motion should be observed, and any impairment of voluntary movement, including dysmetria, athetosis, or dystonia, must be assessed. An acceptable non-invasive test for muscle weakness is clinical observation, such as going up, down the stairs and squat or a child could rise up from the supine position. The manual dexterity examination includes jumping in place, kicking and throwing a ball or hopping on one foot (35).
4.3. Laboratory Experiments
In lack of a potent particular etiology after a detailed history and physical examination, another diagnostic approach is suggested for the physician (40). Cytogenetic analysis in the individual was the first standardized test for the overall evaluation process of mental retardation, especially in children with hereditary anomalies and dysmorphic features. Chromosome aberrations are the only main prevalent known reason that is observed in unselected ID patients. Chromosome analysis can be provided with smaller deletions, duplications, and genetic material, subtle rearrangements, and detection in the child with developmental delays. The mental retardation guideline confirmed that individuals with ID referred without any obvious diagnosis require chromosomal analysis by karyotype 500 resolutions. Furthermore, for those with doubtful symptoms for a microdeletion syndrome, fluorescence in situ hybridization (FISH) test is suggested. The conception of the molecular background and genome localization can be identified as possible rearrangements and translocations in Miller-Dieker, velocardiofacial, and Williams syndromes (41).
The karyotype along with sub-telomeric region analysis can recognize chromosome aberration in approximately 10% of patients with developmental delay (42). Some evidence estimated that the total detection rate of gene anomalies for ID is less than 5% by a karyotype, 5% to 6% by sub-telomeric analysis (FISH) and less than 10% by CGH (comparative genomic hybridization) array (43, 44).
Microarray analysis, including single nucleotide polymorphism or comparative genomic hybridization, can be a helpful method for detection of pathogenic gene/variation in clinical assessment. This analysis has a 100-fold better resolution compared to karyotype (45). Guidelines for the interpretation of microarray analysis are available (46). The primary diagnostic phase is parents testing to define whether the copy number variation (CNV) is de novo and which condition is rather pathogenic. Generally, hereditary CNV of a healthy parent is benign. However, several CNVs have been presented that have uncertain penetrance, hence, careful counseling has been offered (46).
4.4. Whole Exome Sequencing
Despite, development of diagnostic tests, evidence indicates that infrequent de novo point mutations can be the main reason for development delay (47). Often, ID occurred in sporadic forms, without clear etiology. Also, this document creates further support for the de novo mutations hypothesis in ID (16). Although one thousand genes lead to ID yet there is no precise diagnostic method for approximately 50% of ID patients. Elevated recessive disorders frequency was due to consanguineous marriages (48). These types of marriage rate are about 40% in Iran. Homozygosity mapping has proven to be a potent tool for discovering disease-associated genes in consanguineous families. Whole exome sequencing (WES) method is a sequencing and capture technique and can assess 1% of the genome that includes protein-coding data. This technique was primarily performed for gene detection in 2009 (49). Exome enrichment and whole exome sequencing have been presented with a rapid method and cost-effective for universal mutation screening and gene recognition in the coding region of the genomes. Some single candidate genes/variants were recognized for Iranian families (2).
The WES is a promising technique to manifest numerous alterations in the unknown clinical symptom, and can probably be detected in the gene mutation related to the delay-onset disease that is asymptomatic yet similar to breast cancer susceptibility genes (BRCA1 and BRCA2) and Huntington disease. Detailed counselling in two phases of WES (pre and post) could be beneficial for decision in future gestation (50).
5. Main Synaptic Signaling Pathways Effective in Intellectual Disability
Numerous evidence demonstrated that the great functional proteins related to ID are located in synaptic compartments, and novel reports indicated the definitive role of synaptic proteins in dendritic spines, differentiation, and synaptic regulation. Therefore, this study focused on synaptic signaling pathways that involved in synaptopathy (Figure 3) (51, 52).

Schematic synaptic signaling pathways involved in intellectual disability. The presynaptic and postsynaptic molecules that involved in synapse formation, maturation and regulation.
5.1. Rab Pathway
The Rab molecule belongs to GTPases and is efficient in developmental delay through mutations in α GDI moderator and Rab39 GTPase. Some missense mutations of TBC1D24 genes in both major domains have been reported (53). The TBC1D24 by TBC domain can bind to ADP ribosylation factor 6 (ARF6) and down-regulated its function. Moreover, four mutations were observed in IQ-like domains can decrease the GEF act. Over-expression of ARF6 leads to epilepsy in ID patients with TBC1D24 gene mutations (54).
5.2. Ras Pathway
Ras GTPases belonged to a wide group of small G proteins that were effected in ID. SYNGAP1 gene activates the molecule of synaptic GTPase and DLG3/SAP102 gene, which activated SAP102 protein involved in ID (55). Membrane-associated guanylate kinase (MAGUK), including SH3 (Src homology region 3), PDZ (PSD-95/Disc large/Zonula occludes), and GK (Guanylate kinase) domains can bind to microtubule and actin cytoskeletons (56). Some evidence indicated that the SynGAP lead to Racup-regulation and is involved in the morphology of dendritic spine by Ras pathway. Rac/PAK/LIMK pathway can be linked to cofilin and SynGAP through regulation of both members of Rac GEFs (Tiam1 and Kalirin7) (57). Furthermore, ID has a relationship with two signaling pathways in synaptic plasticity (cytoskeleton activity and trafficking in a receptor) (58).
5.3. IL1RAPL1/JNK/PSD-95 Pathway
IL1RAPL1 is a transmembrane molecule with 150 amino acids in the carboxyl-terminal and belongs to a recent IL1/Toll receptor group (59). IL1RAPL1 protein has different mutations linked with ID (non-syndromic) and autism. Up-regulation of IL1RAPL1 in neuroendocrine can decrease the N-type of voltage-gated Ca2+ channel function in presynaptic neurons (60).
5.4. The Rho GTPase Pathway
Rho GTPase has an essential role in actin cytoskeleton dynamic regulation and affected endo and exo endocytosis (61). Ophn1 (oligophrenin1) is the first Rho pathway gene that is involved in ID. Lack of activity and duplication due to mutation of Ophn1 may relate to ID. Control of actin cytoskeleton formation is one of the major activities of the Rho GTPases (62). Furthermore, it is a critical modulator in neuron morphogenesis, such as the spine of dendritic that leads to ID disease (63).
6. Conclusions
Each novel finding of the pathogen genes in the molecular pathway of the brain is critical for physicians and directly affects families. Early diagnosis leads to timely intervention in neuropathies, such as intellectual disability or developmental delays that improve educational intervention planning in the near future. The authors hope these discoveries of de novo point mutations in ID lead to new insights in the molecular pathogenesis and transcriptome map for brightening genetic diagnosis of a disorder in the Iranian population. In addition, finding the cause of novel mutation can lead to opportunities for dividing ID patients with familial and sporadic form, and this may lead to cohort studies, which can benefit targeted therapy. This principle is the major point towards personalized medicine in the affected population.
