

1. Background
Colorectal cancer (CRC) remains a leading cause of cancer-related mortality worldwide, driven by genetic, environmental, and microbial factors that contribute to its progression (1). Mutations in proto-oncogenes, tumor suppressor genes, and DNA repair mechanisms contribute to CRC development, while its molecular heterogeneity complicates precise diagnosis and treatment (2). Identifying predictive and prognostic gene signatures is essential for advancing personalized therapies and improving clinical outcomes (3). The gut microbiota plays a pivotal role in regulating immune responses, metabolism, and cancer progression (4, 5), with dysbiosis frequently linked to CRC tumorigenesis through transcriptomic alteration (6, 7). Modulating the gut microbiome has emerged as a promising approach to enhance treatment efficacy and reduce adverse effects (8). Among microbiota-derived metabolites, phenylacetaldehyde (PAA) exhibits antioxidant, anti-inflammatory, and tumor-suppressive properties (9, 10). While primarily sourced from plant-based diets, gut bacteria also biosynthesize PAA via aromatic amino acids (AAAs) fermentation (11, 12). As a key phenylpropanoid-derived metabolite, PAA disrupts tumor metabolism, affecting phenylalanine-derived pathways critical for cancer cell proliferation (13). It has been implicated in metabolic reprogramming, altering cellular energy dynamics, and restricting tumor growth (14). Mechanistically, PAA modulates oncogenic signaling, exerting its anti-cancer effects through STAT3/IL-6 pathway suppression, oxidative stress induction, and cancer stem cell (CSC) inhibition (14, 15). Given these mechanistic insights, our study seeks to evaluate PAA's transcriptomic impact on CRC, assessing its ability to modulate oncogene expression and tumor-related pathways.
2. Objectives
This study aims to explore PAA’s anti-cancer potential in reducing CRC mortality by leveraging bioinformatics tools and databases to identify downregulated genes. Cancer Genome Atlas (TCGA) data analysis was used to examine expression changes between CRC and normal tissues, and GSE207618 analysis helped pinpoint genes influenced by PAA. Further Cox regression analysis assessed the relationship between gene expression and patient mortality rates. Co-expression network analysis uncovered key molecular pathways linked to prognostic genes, providing deeper insight into PAA’s therapeutic role in CRC progression.
3. Methods
3.1. Data Collection, Normalization, and Differential Expression Analysis
A gene expression omnibus (GEO) study (accession number GSE207618) was used to identify the effect of PAA on CRC-associated genes. In this study, CRC cell lines (HCT-116 and RKO) were treated with PAA, and their influence on transcriptomic changes was analyzed using RNA sequencing. The study included four control samples and five PAA-treated samples. The raw data were downloaded and processed in the RStudio environment. Normalization was conducted using the TMM method, and genes with zero expression were excluded based on the CPM criterion (< 10), using the edgeR package (16). The remaining expression matrix was transformed to a log2 Scale using the limma package and the RMA method for further analysis (17). To determine differential expression, the linear model method was applied to compare gene expression differences between treatment and control groups. Subsequently, the raw format of CRC data (TCGA-COAD) was obtained using the TCGAbiolinks package (18), and TCGA data were normalized using the same method. Differential expression analysis was then performed between 480 colorectal CRC samples and 41 normal samples based on the count expression matrix.
3.2. Displaying Hub Genes Using the Protein-Protein Interaction Network
The protein-protein interaction (PPI) network was utilized to identify hub genes among the downregulated candidate genes affected by PAA treatment. Genes were selected based on differential expression analysis from the GSE207618 dataset, applying logFC < (-1) and FDR < 0.01 as selection criteria. Additionally, the expression levels of these genes were examined in CRC samples from the TCGA dataset, considering logFC > 1 and FDR < 0.01. The interactions among all candidate genes were explored using the STRING tool (http://string-db.org). To further classify and prioritize hub genes, the Cytohubba plugin in Cytoscape (v3.7.2) was employed using the maximal clique centrality (MCC) method. Subsequently, pathways related to hub genes were identified using the MsigDB database via Enrichr (https://maayanlab.cloud/Enrichr/), with a significance threshold of FDR < 0.01. Finally, the Human Protein Atlas was used to assess the protein expression levels of hub genes in CRC, based on immunohistochemistry (IHC) data.
3.3. Survival Analysis and Prognostic
To assess the impact of candidate genes on the survival outcomes of CRC patients, TCGA clinical data were analyzed based on patients’ vital status and overall survival duration (days alive). Gene expression values were normalized using Z-score scaling across all samples, and each Z-score was integrated with clinical data. Cox regression analysis was applied to evaluate the association between gene expression levels and patient mortality rate. Eventually, survival analysis using the Kaplan-Meier curve stratified the results into high- and low-expression groups, based on the median expression levels of candidate genes in CRC samples.
3.4. Co-expression Networks
The co-expression network was depicted using the normalized expression matrix from the TCGA database to determine pathways associated with poor-prognosis genes in CRC. Hence, the correlation between the expression levels of candidate genes and all other genes in the expression matrix was examined using the Pearson correlation test. Co-expressed genes were selected based on the criteria R > 0.5 and P < 0.01. Finally, pathways connected to correlated genes within the co-expression network were found using the MsigDB database via Enrichr.
3.5. Software and Statistical Analysis
All data processing and analysis were conducted using the R programming language (v4.0.2). Differential expression between groups was assessed using the linear model method, while the LogRank test was applied to compare survival distributions between CRC and normal samples. Furthermore, GraphPad Prism (v8) was utilized for graphical visualization, and Cytoscape (v3.7.2) was employed to construct and visualize PPI and co-expression networks.
4. Results
4.1. Identification of Genes Downregulated by Phenylacetaldehyde Treatment
The GSE207618 study was employed to identify genes affected by PAA treatment. Differential expression analysis revealed a significant decrease in the expression of 107 genes in CRC cell lines treated with PAA (LogFC < (-1), FDR < 0.01). Among these downregulated genes, 54 genes exhibited overexpression in CRC samples compared to normal ones, based on TCGA data analysis (LogFC > 1, FDR < 0.01, Figure 1A). These findings suggest that PAA may have the potential to regulate the altered expression of these 54 identified genes in CRC (Figure 1B).
 that are downregulated by phenylacetaldehyde (PAA). A, a heatmap illustrating Cancer Genome Atlas (TCGA) data analysis results, highlighting the overexpression of 54 candidate genes in cancer samples (n = 480) compared to normal samples (n = 41); B, a heatmap depicting expression changes of 54 candidate genes following PAA treatment in CRC, based on GSE207618 data analysis.")
Identifying overexpressed genes in colorectal cancer (CRC) that are downregulated by phenylacetaldehyde (PAA). A, a heatmap illustrating Cancer Genome Atlas (TCGA) data analysis results, highlighting the overexpression of 54 candidate genes in cancer samples (n = 480) compared to normal samples (n = 41); B, a heatmap depicting expression changes of 54 candidate genes following PAA treatment in CRC, based on GSE207618 data analysis.
4.2. Discovery and Functional Characterization of Hub Genes
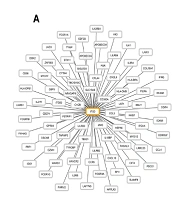
The PPI analysis identified that among the 54 candidate genes, 32 genes exhibited significant interactions (Figure 2A). The MCC criterion determined that nine key genes — MCM2, MCM4, MCM6, MCM7, MCM10, PCNA, UHRF1, FEN1, and KIF11 — function as hub genes, demonstrating the highest levels of interaction (Figure 2B). To gain deeper insights into the functional role of these hub genes, the Enrichr tool was utilized. Analysis using the MsigDB database displayed their involvement in pathways such as mTORC1 signaling, DNA repair, G2-M checkpoint, Myc targets V1, and E2F targets, all of which play critical roles in tumor progression, angiogenesis, and metastasis (Figure 2C FDR < 0.01). Furthermore, our results indicate that hub gene expression levels are significantly elevated in cancer compared to normal samples from TCGA-COAD data (Figure 3A). However, PAA downregulates their expression in CRC cells, as evidenced by GSE207618 data analysis (Figure 3B). Additionally, findings from the Human Protein Atlas confirm high protein levels of hub genes in CRC patients (Figure 3C). Thus, PAA may exert a significant suppressive effect on CRC by reducing hub gene expression, highlighting its potential as a therapeutic agent.
 network of downregulated genes under phenylacetaldehyde (PAA) treatment in colorectal cancer (CRC); B, maximal clique centrality (MCC) score highlighting hub gene relevance; C, enrichment results identifying key pathways linked to hub genes within the PPI network.")
Selection of hub genes and analysis of their relevant pathways. A, protein-protein interaction (PPI) network of downregulated genes under phenylacetaldehyde (PAA) treatment in colorectal cancer (CRC); B, maximal clique centrality (MCC) score highlighting hub gene relevance; C, enrichment results identifying key pathways linked to hub genes within the PPI network.
. A, significant changes in hub gene expression in cancer samples compared to normal, based on the Cancer Genome Atlas (TCGA) dataset analysis; B, the effect of phenylacetaldehyde (PAA) treatment on decreasing hub gene expression in CRC cell lines, demonstrated through GSE207618 data analysis (**** P < 0.0001); C, immunohistochemistry (IHC) data illustrating hub gene expression levels in CRC, as reported in the Human Protein Atlas database.")
Analysis of hub gene expression alteration in colorectal cancer (CRC). A, significant changes in hub gene expression in cancer samples compared to normal, based on the Cancer Genome Atlas (TCGA) dataset analysis; B, the effect of phenylacetaldehyde (PAA) treatment on decreasing hub gene expression in CRC cell lines, demonstrated through GSE207618 data analysis (**** P < 0.0001); C, immunohistochemistry (IHC) data illustrating hub gene expression levels in CRC, as reported in the Human Protein Atlas database.
4.3. Phenylacetaldehyde’s Potential in Suppressing the Expression of Poor-Prognostic Genes
The impact of PAA on CRC patient survival was analyzed using TCGA clinical data. Cox regression analysis of downregulated genes by PAA suggested that increased expression of two genes, HSPA1B and IFI30, in CRC samples was associated with a higher risk of cancer mortality. These genes, recognized as poor-prognosis markers, can serve as potential indicators in CRC patients (HR > 1 and logRank < 0.05). To further confirm this, gene expression levels were divided into low- and high-expression groups using Z-score, followed by Kaplan-Meier survival analysis for IFI30 (Figure 4A) and HSPA1B (Figure 4B). To better define the role of these poor-prognosis genes in CRC development and malignancy, their expression levels were examined in CRC and normal tissue samples from the TCGA database. Results indicated a significant increase in IFI30 (Figure 4C) and HSPA1B (Figure 4D) expression in CRC samples. Conversely, PAA was found to markedly reduce the expression of IFI30 and HSPA1B, as evidenced by GSE207618 data analysis. These findings highlight PAA’s potential to mitigate CRC mortality by modulating the expression of malignancy-associated genes.
 patient mortality by phenylacetaldehyde (PAA) treatment. A and B, Kaplan-Meier plots depicting IFI30 and HSPA1B as poor-prognostic genes (HR > 1); C and D, differential expression analysis of IFI30 and HSPA1B in TCGACOAD cancer samples compared to normal; E and F, the impact of PAA treatment on downregulating IFI30 and HSPA1B expression.")
Modulation of gene expression associated with colorectal cancer (CRC) patient mortality by phenylacetaldehyde (PAA) treatment. A and B, Kaplan-Meier plots depicting IFI30 and HSPA1B as poor-prognostic genes (HR > 1); C and D, differential expression analysis of IFI30 and HSPA1B in TCGACOAD cancer samples compared to normal; E and F, the impact of PAA treatment on downregulating IFI30 and HSPA1B expression.
4.4. Correlation Between Poor-Prognostic Gene Expression and Key Carcinogenesis Pathways
Pathways related to IFI30 and HSPA1B were identified through the co-expression network. Our investigation determined that the expression levels of IFI30 (Figure 5A) and HSPA1B (Figure 5C) are significantly correlated with 80 and 22 other genes, respectively, with a correlation coefficient greater than 0.5 and P < 0.01. Enrichment analysis indicated that genes correlated with IFI30 are involved in pathways related to interferon-gamma response, IL-6/JAK/STAT3 signaling, IL-2/STAT5 signaling, interferon-alpha response, TNF-alpha signaling via NF-kB, KRAS signaling up, and inflammatory response (Figure 5 FDR < 0.01). Similarly, co-expressed genes with HSPA1B participate in various cancer-related pathways, including TNF-alpha signaling via NF-kB, hypoxia, p53 pathway, apoptosis, mTORC1 signaling, epithelial-mesenchymal transition, glycolysis, Myc targets V2, PI3K/AKT/mTOR signaling (Figure 5B FDR < 0.01). These findings suggest that IFI30 and HSPA1B may contribute to CRC pathogenesis through these pathways.

The correlation between cancer-related pathway genes and poor prognosis genes. A, co-expression network displaying genes correlated with IFI30 expression; B, enrichment results for all genes within the IFI30 co-expression network; C, co-expression network visualizing genes correlated with HSPA1B expression; D, significant pathways associated with genes in the HSPA1B co-expression network.
5. Discussion
The PAA, a microbiota-derived metabolite, has shown anticancer properties, including tumor suppression in breast cancer by inhibiting proliferation, inducing apoptosis, and impairing tumor progression (19). Biosynthesized in the colon through microbial fermentation of AAAs (11), PAA may play a key role in CRC suppression. This study provides evidence that PAA influences the CRC transcriptome, exerting an inhibitory effect on tumor development. Key hub genes MCM2, MCM4, MCM6, MCM7, MCM10, PCNA, UHRF1, FEN1, and KIF11 were identified through PPI network analysis, showing significant overexpression in CRC. Transcriptomic analysis of TCGA and GSE207618 datasets confirmed PAA’s ability to downregulate these oncogenes, reinforcing its anti-cancer potential. Pathway enrichment analysis linked these genes to tumor mutation, invasion, and proliferation, further supporting PAA as a promising therapeutic modulator in CRC.
This study also identifies IFI30 and HSPA1B as poor-prognosis oncogenes in CRC, linking IFI30 to immune response and HSPA1B to cell proliferation. IFI30 has been recognized as a prognostic biomarker across multiple malignancies, playing a key role in tumor immunoregulation through antigen processing and immune cell infiltration (20). Its expression is related to tumor-associated macrophages, making it a potential target for immunotherapy (21). Similarly, HSPA1B is linked to poor prognosis in various cancers and contributes to tumor progression, metastasis, and therapy resistance. Overexpression of HSPA1B in CRC correlates with disease severity and treatment response, suggesting its potential as a biomarker (22). Targeting HSPA1B-mediated stress pathways may enhance chemotherapy or immune checkpoint inhibitor efficacy (23). Transcriptomic analysis of TCGA data confirmed IFI30 and HSPA1B overexpression in CRC, while GSE207618 analysis revealed PAA-induced downregulation, reinforcing PAA’s therapeutic role in mitigating tumor malignancy.
To improve the relevance of our findings, we compared the identified genes with established CRC gene signatures, leveraging findings from TCGA, GEO, and large-scale transcriptomic studies. IFI30 and HSPA1B are linked to tumor immunoregulation and stress-response pathways, frequently associated with poor prognosis in CRC (22, 24). Their overexpression aligns with prior studies identifying immune-modulating factors as potential biomarkers. The MCM family and PCNA, as cell cycle regulators, are crucial for tumor proliferation and chemoresistance (25, 26). Their downregulation by PAA reinforces its therapeutic relevance in targeting proliferative pathways. UHRF1 and FEN1, involved in DNA repair and chromatin remodeling, contribute to tumor progression and metastasis (27, 28). The PAA’s ability to reduce their expression suggests a role in altering the tumor’s epigenetic profile, consistent with previous CRC transcriptomic models. KIF11, a key regulator of tumor invasion and metastasis, has been consistently identified in CRC-specific gene signatures (29). The PAA’s suppression of KIF11 expression further supports its potential in restricting tumor cell motility and invasion. Overall, this comparative analysis underscores PAA’s ability to modulate oncogenic pathways, aligning with established CRC molecular targets, and reinforcing its potential as a novel therapeutic candidate.
This study highlights PAA’s therapeutic potential in CRC, but several limitations must be acknowledged. The analysis relies solely on in silico methods, using public transcriptomic datasets without experimental validation. The lack of in vitro and in vivo studies restricts direct confirmation of PAA’s biological effects on tumor progression. Additionally, variability in dataset sample sizes may introduce biases in gene expression trends, emphasizing the need for controlled experimental validation. To advance PAA as a clinical microbiota-derived anti-cancer therapy, several research areas must be explored: In vitro validation using cell culture assays is essential to assess its effects on CRC cell viability, apoptosis, and gene expression; in vivo studies with animal models will provide preclinical insights into PAA’s therapeutic potential in CRC suppression.
5.1. Conclusions
This study highlights the potential of PAA, a microbiota-derived metabolite, in suppressing CRC progression. Our findings demonstrate that PAA exerts significant oncogene downregulation, suggesting its role in modulating tumor development and malignancy. Given its capacity to inhibit key cancer-associated pathways, PAA presents a promising avenue for therapeutic exploration. Further clinical and experimental investigations are needed to validate its efficacy, paving the way for microbiome-based interventions in CRC treatment.
