








1. Background
Clinical features of megalencephalic leukoencephalopathy with subcortical cysts (MLC) are usually characterized by early-onset macrocephaly during the first months of life and diffuse cerebral white matter abnormalities. Gradually this clinical picture is continued by slowly progressive motor and mild mental decline, ataxia, spasticity and seizures (1). However, macrocephaly may be variable among patients ranging 4 to 6 year standard deviation over the mean. On the other hand, some features such as seizure are possibly observed in a number of patients (2).
Mutations of MLC1 (OMIM 605908) and GLIALCAM genes (OMIM 611642) are responsible for 75% and 20% of affected individuals, respectively (1, 3, 4).
MLC1 is located on chromosome 22q13.33 and expressed mainly in the brain. It encodes an oligomeric plasma membrane protein in contact regions of astrocyte (5). The structure of MLC1 protein similar to ion channels may transport ions through astrocyte membrane, although its exact role remains undiscovered (6).
Intronic splice site mutations are assumed to disrupt the function of gene according to ACMG standards and guidelines and accounts for about 4.8% of MLC1 mutations (http://www.hgmd.cf.ac.uk/ac/search.php) (7).
2. Objectives
In this study, a multi affected family with variable phenotypes including a normal adult individual is reported to illustrate the wide clinical spectrum of MLC. In addition, an update of MLC1 splice site mutations is presented and the previously adult cases with splice site mutations are also reviewed and compared.
3. Methods
3.1. Clinical Report
3.1.1. Case Presentation
Here we evaluated a family with 4 affected members with heterogeneous clinical manifestations. The clinical and MRI data were documented for all these 4 patients. A comprehensive search was conducted on clinical variations of MLC with focus on splice site mutation in MLC1 gene. The informed consent was signed by parents and legal guardians of participants. Ethics Committee of Children’s Medical Center hospital approved the study.
3.1.1.1. Index Case
A seven-year-old boy was referred to Myelin Disorders Clinic at Children’s Medical Center (CMC), Tehran, Iran. His complaints were macrocephaly and gait disturbance. He was the third child of healthy non-consanguineous parents. Birth weight and head circumference were not available.
According to their parents, his developmental milestones were within normal range (Sitting alone at 7 months, rolling over at 8 months, start speaking few words and walking at 18 months). The first symptom of the patient was noticed by parents at the age of 2 year when he showed right leg clumsiness and progressive difficulty in walking. He experienced frequent falls after starting to walk few steps in the age of 2.5 year. He was admitted to hospital for medical evaluation but no definite diagnosis was achieved. After that he had no medical follow-up until age of 7 years. At age of 7, he referred to Myelin Disorders Clinic due to unsteady gait, progressive enlarged head circumference, learning disorder and abnormal school performance. He had never experienced any seizure. In developmental evaluation of index case, speech domain was acceptable but mild cognitive impairment and behavioral abnormalities were detected. Neurologic examination revealed macrocephaly with head circumference of 56 cm (> 99 percentile for age and sex), lower limbs spasticity and clumsy gait but no joint contracture. Brisk tendon reflexes at the lower limbs (3/4) were noticed whereas it was normal in the upper limbs. Brain magnetic resonance imaging (MRI) was done due to suspicion of a neurodegenerative disorder. Axial T2-weighted, FLAIR and T1-weighted imaging of brain MRI demonstrated diffuse abnormal white matter signals, high in T2-W and low in T1-WI images in favor of demyelination type leukodystrophy. Axial FLAIR images at the level of temporal lobes showed clearly small subcortical cyst formations (Figure 1A-F). Molecular study was performed according to MRI findings to confirm the diagnosis. At the last visit, the patient could walk without aid more than 10 meters but with some degree of imbalanced gait, mild dysarthria, mild school dysfunction and had no seizure.
. C - E, MRI, axial T2 WI, FLAIR and T1 WI images at the level of frontoparietal centrum semiovale demonstrate diffuse abnormal white matter signals, high in long TR sequences and low in T1-WI in favor of demyelination type leukodystrophy. F, MRI, coronal T2WI at the level of basal ganglia display diffuse subcortical and deep white matter involvement.")
A, brain MRI of the index patient at age of 7 years old, axial T2 WI, at the level of temporal lobes reveals abnormal symmetric hyper signal white matter. B, MRI, axial FLAIR at the same level shows clearly small subcortical cyst formations at the right and the left anterior temporal lobes (white arrows). C - E, MRI, axial T2 WI, FLAIR and T1 WI images at the level of frontoparietal centrum semiovale demonstrate diffuse abnormal white matter signals, high in long TR sequences and low in T1-WI in favor of demyelination type leukodystrophy. F, MRI, coronal T2WI at the level of basal ganglia display diffuse subcortical and deep white matter involvement.
3.1.1.2. Case 2
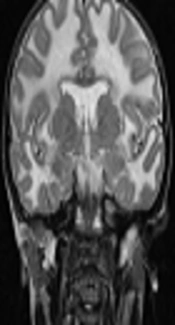
During the family segregation analysis, we evaluated the mother of case 1 more precisely. She underwent more medical evaluation by a retrospective manner when we found the same bi-allelic genetic abnormality of proband (case 1) in her genetic test results. She was a 45-year-old female, the third child of healthy non-consanguineous parents. She had never experienced any seizure. Her gait and cognitive skills were completely normal. In physical and neurologic examination, her head circumference was 57 cm. No spasticity was detected in all four limbs and plantar response was flexor. Other parts of neurological examination were also normal except for increased deep tendon reflexes in lower limbs at both knee and ankle joints (3/4). Brain MRI was done due to presence of the same bi-allelic mutation of index case in mother which surprisingly revealed diffuse abnormally increased white matter signal in axial and coronal T2-weighted imaging at the level of basal ganglia in addition to bilateral anterior temporal subcortical cysts at the level of temporal lobes in axial FLAIR sequence (Figure 2A-D).
. B, Coronal T2 WI at the level of basal ganglia display diffuse subcortical and deep white matter involvement and anterior temporal subcortical cysts (white arrow). C and D, MRI, axial T2 WI and FLAIR at the level of frontoparietal centrum semiovale reveals diffuse abnormal high signal white matter and some widening of sulci.")
A, Brain MRI of the patient’s mother at age of 45 years old, axial FLAIR, at the level of temporal lobes shows abnormal increased white matter signal and bilateral anterior temporal subcortical cysts (white arrows). B, Coronal T2 WI at the level of basal ganglia display diffuse subcortical and deep white matter involvement and anterior temporal subcortical cysts (white arrow). C and D, MRI, axial T2 WI and FLAIR at the level of frontoparietal centrum semiovale reveals diffuse abnormal high signal white matter and some widening of sulci.
Other affected family members. In the studied family, we found that two aunts of the proband are also affected. One of the aunts was 33 years old with a history of falling down at age 7, after which she developed motor regression, mild cognitive disability and few seizures in addition to hand tremor. The other aunt was 30 years old with also a history of falling down at age 8, thereafter she developed gross and fine motor regression during the following months without any seizure and mild cognitive disability. They were not available for exact clinical examination. Figure 3 shows pedigree of the affected family.

Pedigree of affected family. The proband, his mother and two aunts were homozygous. Other members of family carried heterozygous mutation.
3.1.2. Genetic Testing, Data Extraction
Blood samples were taken from proband and other family members. DNA extraction was performed using salting out method. Primers were designed for all 12 exons and exonic-intronic boundaries of MLC1 gene. Accession numbers of the reference sequence are NM-015166.3 and NG-009162.1.
All exons and exonic-intronic boundaries of MLC1 gene were amplified using PCR. Amplification reactions were performed in a final volume of 50 µL containing 10 pmol of each primer, 150 ng template DNA, 25 µL Taq DNA polymerase 2× master mix red containing 1.5 mM MgCl2, 0.4 mM dNTPs, 0.2 units/µL Ampliqon Taq DNA polymerase and carried out for 30 cycles: denaturation at 95°C for 30 seconds, annealing at 62°C for 30 seconds, extension at 72°C for 30 seconds, followed by a final extension for 5 minutes at 72°C. Briefly, DNA sequencing was performed on the PCR products using the BigDye method by sequencing analyzer of ABI 3500XL model (PE Applied BioSystems, Massachusetts, USA).
A comprehensive search was done through PubMed, John Wiley, Springer and Human Gene Mutation Database (HGMD) databases using the following keywords: MLC1, Mutations and/or intronic mutations.
4. Results
4.1. Genetic Findings
Genetic study revealed an intronic splice-site variant in intron 2 (c.177+1G>T) of MLC1 gene in four members of this family as a homozygous variant. This variant was heterozygous in proband’s father but surprisingly homozygous in his normal mother. Both his affected aunts were homozygous for c.177+1G>T and other aunts, uncles and his siblings were heterozygous for this variant. The c.177+1G>T was considered to be disease-causing by Mutation Taster. Up to now, 33 patients with twenty MLC1 splice-site mutations with an age range of 4 months to 44 years have been reported to cause MLC; 5 (25%) of whom had c.177+1G>T mutation (Table 1).
| No | Mutation | Nucleotide Position | Chromosome Location | Gender | No. of Patients | Inheritance (Zygosity) | Age of Onset | Population | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 1 | IVS2+1G>T | c.177+1G>T | chr22:50523154C>A | M | 1 | Homozygous | 1.5 y | Iran | This study |
| F | 2 | Homozygous | 4 mo | Iran | (1, 2) | ||||
| M | 1 | Homozygous | 38 y | Turkey | (3) | ||||
| M | 1 | Homozygous | 8 mo | Italy | (4) | ||||
| 2 | IVS7-2A>C | c.598-2A>C | chr22:50512763T>G | M | 1 | Heterozygous | 5 mo | Chinese | (5) |
| Fetus, F | 2 | Heterozygous | Chinese | (6) | |||||
| 3 | IVS5+1G>A | c.423+1G>A | chr22:50518346C>T | - | 1 | Heterozygous | 9 mo | Italy | (4) |
| M | 1 | Homozygous | 5 mo | Iran | (2) | ||||
| - | 1 | Homozygous | - | Africa | (7) | ||||
| 4 | IVS9-1G>C | c.772-1G>C | chr22:50506985C>G | M | 1 | Heterozygous | 8 mo | Chinese | (8) |
| F | 1 | Heterozygous | 4 mo | ||||||
| F | 1 | Homozygous | 7 mo | ||||||
| 5 | IVS4-1G>A | c.322–1 G>A | chr22:50518449C>T | M | 1 | Heterozygous | 6 mo | USA | (9) |
| 6 | IVS7+1G>A | c.597+1G>A | chr22:50515269C>T | M | 1 | Heterozygous | 6 mo | USA | (9) |
| 7 | IVS10-226 T>G | c.895-226 T>G | chr22:50502893A>C | F | 1 | Homozygous | 44 y | Italy | (10) |
| 8 | IVS2-10T>A | 178-10T>A | chr22:50521612A>T | - | 1 | Heterozygous | - | Croatia | (11) |
| 9 | IVS3-1 G>A | 268-1G>A | chr22:50518827C>T | - | 1 | Heterozygous | - | USA | (11) |
| 10 | IVS5+6T>G | c.423+6T>G | chr22:50518341A>C | M | 2 | Heterozygous | 6 mo | Turkey | (4) |
| 11 | IVS8+1G>A | c.714+1G>A | chr22:50512644C>T | - | 1 | Heterozygous | - | Croatia | (11) |
| 12 | IVS10-1G>C | c.895-1G>C | chr22:50502628C>G | M | 1 | Heterozygous | 8 mo | Italy | (4) |
| 13 | IVS10-2A>G | c.895-2A>G | chr22:50502629T>C | M,F | 2 | Homozygote | - | Turkey | (12) |
| 14 | IVS11-2A>G | c.1060-2A>G | chr22:50500088T>C | - | 1 | Heterozygote | - | Japan | (12) |
| 15 | IVS3+1G>C | c.267+1G>C | chr22:50521512C>G | M | 1 | Homozygote | - | Turkey | (13) |
| 16 | IVS4-2A>G | c.322-2A>G | chr22:50518450T>C | M | 1 | Homozygote | 7 y | Turkey | (13) |
| 17 | IVS5+6T>C | c.423+6T>C | chr22:50518341A>G | M | 1 | Heterozygote | - | Turkey | (13) |
| 18 | IVS4-6T>C | c.322-6T > C | chr22:50518454A>G | F | 1 | Homozygous | 11 mo | Iran | (14) |
| 19 | IVS10-1G>A | c.895-1G>A | chr22:50502628C>T | M | 1 | Homozygous | 3 y | Chinese | (5) |
| 20 | IVS513insT | c.423+2dup | chr22:50518344_50518345insA | - | 1 | Compound | - | Indian | (12) |
| M | 1 | Heterozygote | China | (15) |
5. Discussion
Considerable heterogeneity in clinical features of MLC is seen and the onset age of clinical pictures ranges from birth to 25 years, with a median age of 6 months (16). The maximum age of clinical presentation has been reported in a 52-year-old patient with 6 years history of seizures (11). According to finding an asymptomatic adult individual in a familial case series of MLC due to a pathogenic splice site mutation in MLC1, we conducted a comprehensive search on variants of MLC1 gene to find all reported splice site variants and their clinical spectrum. Until now 20 splice site mutations in MLC1 gene have been reported to cause MLC (Table 1).
Macrocephaly, lower limbs spasticity, cognitive impairment and behavioral abnormalities without seizure were the main clinical features of the proband case, in addition to white matter involvements but detailed neurological examination on probond’s mother revealed she was normal except for increased deep tendon reflexes in lower limbs at both knee and ankle joints (3/4). Her head circumference was 57 cm that seems to be in borderline range of macrocephaly for women (17). Nevertheless, diffuse white matter involvements and anterior temporal subcortical cysts were found in mother; she was also homozygous for the pathogenic variant of the proband case. The clinical scenario of two aunts was compatible with the usual course of MLC and molecular evaluation confirmed the diagnosis.
The first report of an oligosymptomatic adult patient was a 38 year old Turkish male having homozygous c.177+1G>T with no major complication related to MLC. He had mild spastic and ataxic gait. MRI of the brain showed diffuse involvement of white matter with temporal subcortical cysts (3). A 37-year-old macrocephalic woman has been introduced with ataxia and spasticity in her legs. Her brain MRI showed multiple subcortical cysts in anterior-temporal and parietal regions (14). Another adult patient related to a male aged 35 years presented with walking difficulties which began at age 6 year and developed progressive gait problem. After that he had experienced loss of sensation, pain and weakness in his upper extremities. His head circumference was 64. He was diagnosed as a case of MLC at the age of 35 years (18). The maximum age of clinical presentation due to splice site mutation in MLC1 has been reported in a 52-year-old patient with 6 years’ history of seizures following which she developed acute deterioration of consciousness, decreased speech output and difficulty leading to rapid neurological deterioration in walking with malignant transformation of cysts in her brain MRI in temporal lobes area. Her head circumference was 57.5 cm (19). An intronic variant, IVS10-226 T>G, has been reported in a mildly symptomatic woman aged 44 years. She presented with macrocephaly, psychomotor development, mild cognitive problem in school and subsequently episodes of seizures during adolescence. Brain MRI was compatible with MLC. MLC1 cDNA analysis revealed a transcript reduction due to this homozygous deep-intronic mutation (10). In addition, a 51 year old oligosymptomatic Japanese patient has also been described with mild cognitive and memory deficit, seizures and weakness of the extremities due to a missense mutation, p. Ser93Leu, but not splice site variant of MLC. The first symptom was cognitive impairment when he was 41 years old (20).
As mentioned above, several adult cases of MLC have been described that showed atypical or mild symptoms due to MLC1 mutation; but there is not any report of clinically asymptomatic patient. This is the first report of an asymptomatic MLC patient from a non-consanguineous family with a pathogenic mutation in MLC1. Our adult case showed no subjective clinical symptoms during two years follow up until 47 years of age. Based on this finding we could expand the spectrum of MLC clinical symptoms and claim that MLC is a rare neurological disease that could be clinically asymptomatic. However, it should be noted that in index case’s mother, symptoms may occur in subsequent years. There is no definite treatment for MLC and it is mostly managed with supportive care and rehabilitation programs such as occupational and physical therapy. Since our adult case did not show any clinical symptoms, she did not receive any of these supportive cares.
In our study, c.177+1G was homozygous in a non-consanguineous marriage; the carrier frequency of mutation should be high in this scenario, as we previously reported some mutations in other genes have a high frequency but others have not been observed in our populations. For example eight common mutations have been observed in CYP21A2 gene while a common mutation in GJB6 in European countries is not observed in our country (21, 22). Considering this point is very important in screening strategies. c.177+1G as the first nucleotide of intron 2 mutated to T leads to disruption of slicing. Exon skipping and/or intron retention are the probable consequences of this substitution which both of them affect the protein structure and function. This mutation showed apparently reduced penetrance or more strongly variable expressivity in this family from a classic presentation of MLC in a child (index case) to an asymptomatic adult individual (index case’s mother). Altogether, previous data shows this mutation has a high penetrance; clinical variability within this family might be attributed to genetic modifiers (17). There are some probabilities about the subclinical phenotype of the individuals carrying this mutation. An unknown protein may exist in these homozygous individuals that could compensate lack of MLC protein. In the same way, when c.177+1G>T occurs, the conserved dinucleotide GT is changed to TT at the beginning of intron, in splicing process an alternative protein may recognize and splice it correctly and a normal mRNA is produced. Any isoform of MLC1-interacting proteins may have a different interaction with this protein so that the function of mutated MLC1 is compensated and the phenotype becomes approximately normal and/or the patient is underdiagnosed. However, in some cases we should look at the variant in context of genetic variations as a whole. Next generation sequencing technologies could provide all genetic variations of patients. Consequently, although MLC is a rare neurodegenerative disease, some cases may be underdiagnosed.
5.1. Conclusions
This studied family provides a clue to identify genetic modifiers of MLC as we here presented an asymptomatic adult case. Finding these genetic agents sheds light on the therapeutic options for the disease so that symptoms of MLC could be ameliorated in presence of these genetic modifiers. Further studies are required to discover these modifiers.
