





1. Background
Thalassemia, one of the most prevalent single-gene hereditary disorders worldwide, is caused by a mutation or deletion of the alpha-globin and beta-globin genes in the human body, causing the globin peptide chain to become imbalanced and resulting in hemolytic anemia, also known as marine anemia. Thalassemia is widespread, with higher rates in the Mediterranean, the Middle East, and Southeast Asia (1). According to the reduction of the peptide chain, thalassemia is classified as alpha-thalassemia (α-thalassemia), beta-thalassemia (β-thalassemia), gamma-thalassemia (γ-thalassemia), delta-thalassemia (δ-thalassemia), and delta-beta-thalassemia (δβ-thalassemia). The α-thalassemia and β-thalassemia are both frequent in the clinic. According to the clinical classification, thalassemia is categorized as mild, moderate, or severe.
The intestinal flora is associated with numerous metabolic pathways in the body and affects human health and differences in the composition and abundance of the intestinal flora when the body is ill. Intestinal flora’s variety, stability, and balance play an important role in maintaining human health in a steady state. Clinically, women with mild thalassemia can have normal pregnancies, delivery, and newborns, just like healthy women. Because individuals with mild thalassemia have no evident clinical signs, their health is essentially indistinguishable from that of the general population. Due to various physiological changes of pregnant women, great physiological and immune function changes will happen in the intestinal flora (2). The intestinal flora might be dysfunctional, resulting in maternal maladjustment, fetal development restriction, and spontaneous abortion (3).
2. Objectives
Metagenomic next-generation sequencing (mNGS) is a technology based on metagenomics that directly analyzes microbial deoxyribonucleic acid (DNA) or ribonucleic acid in samples without growing them. In this study, metagenomics sequencing technology was used to analyze intestinal flora changes in healthy pregnant women and pregnant women with mild thalassemia; the specific species categories were analyzed to identify key bacterial species and assess potential effects on pregnant women and fetuses.
3. Methods
3.1. Research Subjects
All the samples were from pregnant women hospitalized in the Affiliated Hospital of Putian University, Fujian, China. The hospitalized pregnant women in this study took the designated food at the specified time as expected 2 weeks before sampling and did not follow other diets and sampling at a fixed time. The selected pregnant women all came from similar backgrounds, including (1) from nearby suburbs with comparable living habits (e.g., eating habits, living hours, and labor intensity), (2) no family history of other genetic illnesses, and (3) within the age and weight ranges of 26 - 29 years and 55 - 60 kg, respectively. Six healthy pregnant women (control group), seven pregnant women with mild α-thalassemia (treatment B group), and seven pregnant women with mild β-thalassemia (treatment C group) were all subjected to rigorous thalassemia gene testing. The inclusion criteria were (1) no antacids, probiotics, antibiotics, or micro-antibiotics taken by the patients in the previous month, (2) no diabetes or other metabolic diseases influencing the intestinal flora, and (3) without digestive gut surgery. The exclusion criteria were (1) hematochezia symptoms and (2) digestive disorders that might create an imbalance in the intestinal flora.
3.2. Fecal Collection and DNA Extraction
The fresh morning feces of the selected pregnant women were collected and stored at -80ºC immediately. The sable stool samples were extracted with the HostZERO microbial DNA kit (Zymo Research, California, USA). The DNA was tested for quality control and quantified before library construction. The purity and integrity of the DNA were determined using agarose gel electrophoresis, and DNA concentration was measured using a Qubit 3.0 Fluorometer (Invitrogen, USA). Each sample’s DNA fragments ranged from 3000 to 5000 bp, reaching the parameters for creating the DNA library.
3.3. Library Preparation and Metagenomics Sequencing
During library construction, eligible DNA samples were randomly digested into 500-bp-long pieces using an ultrasonic crusher (Covaris, UK). The fragments were ligated to adapters after being end-repaired and A-tailed. Following library preparation, an initial quantification was performed using a Qubit 3.0 Fluorometer, and the library was diluted to 2 ng/µL. Following that, an Agilent 2000 Bioanalyzer (Agilent, USA) was used to assess whether the library’s insert sizes corresponded to expectations. A quantitative polymerase chain reaction was used to quantify the library’s effective concentration (> 3 nM) to ensure library quality. The follow-up metagenome sequencing was completed by Shenzhen Weishengtai Technology Co., Ltd, Shenzhen, China. The Illumina NovaSeq 6000 platform (Illumina, USA) was used for sequencing.
3.4. Quality Control and Genome Assembly
The low-quality sequences in the raw data were firstly removed using Trimmomatic 0.36 software (parameter: ILLUMINACLIP: adapters_path:2:30:10 SLIDINGWINDOW:4:20 MINLEN:50) to ensure the accuracy of subsequent analysis results (4), followed by Bowtie 2 software (parameter: --very-sensitive) to remove the host genome (hg38) to obtain the clean data of the samples (5). The clean data were gained following these filtering procedures, and MEGAHIT software (parameter: --k-list 21,29,39,59,79,99,119,141 --min-contig-len 500) was used for assembly analysis (6). The scaffolds were severed at the N-junctions to produce N-free sequence segments known as scaftigs (7). Bowtie 2 software (parameters: -end-to-end, -sensitive) was used to match the clean data for each sample to the scaftigs of each sample to get PE (paired-end (PE)) readings. MEGAHIT software was used for mixed assembly after pooling the clean readings from each sample, and the remaining assembly parameters were similar to those used for single sample assembly. The fragments of fewer than 500 bp were filtered out of the scaftigs generated by both single sample assembly and mixed assembly (8), and then gene predictions were carried out.
3.5. Gene Prediction, Abundance Analysis, and Species Annotation
Prodigal software (parameters: -p meta) was used to predict open reading frames (ORFs) using the scaftigs for each sample assembly and mixed assembly (≥ 500 bp) (9). The fragments shorter than 100 nt were removed from the prediction results. CD-HIT software (parameters: -G 1 -c 0.9) was used to eliminate redundancy from each sample’s ORF prediction findings, providing an initial non-redundant gene catalog. Salmon software (parameters: -validate Mappings - meta) was used to compare the clean data into the de-redundant genes and determine their relative abundance reads per million of the de-redundant genes (10). Kraken 2 software (parameters: --confidence 0.2) was used to compare the unigenes to the sequences retrieved from the NCBI NR database of bacteria, fungi, archaea, and viruses (11). The lowest common ancestor (LCA) technique was used to assign the categorization level before the first branch as the sequence’s species annotation information. The abundance information of each sample at each categorization level (i.e., genus and species) was collected using the LCA annotation findings and the gene abundance table. The abundance of a species in a sample was calculated as the sum of the gene abundance of the annotated species.
3.6. Statistical Analysis
The collected data were analyzed by SPSS software (version 20.0) and represented by mean±standard deviation. Analysis of variance and Duncan’s multiple-comparison test were used for the comparison between the groups. All the results were considered statistically significant at a P-value of less than 0.05. The Wilcoxon rank-sum test was used to detect significant intergroup differences in the relative abundance of taxonomy and species features (P < 0.05).
4. Results
4.1. Quality Control of Metagenomics Sequencing
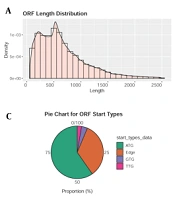
The clean reads of each sample account for more than 92% of the raw reads after processing the original sequencing data. Typically, there are numerous redundant sequences in the ORF sequences of predicted genes, making it simple to commit errors and skew the findings. CD-HIT software (12) was applied to the cluster for removing redundant sequences (cluster similarity ≥ 0.95; coverage ≥ 0.9). The longest ORF sequence from each category was chosen as the representative sequence to construct a set of non-redundant gene sequences (Figure 1).
 sequence; B, Predicting the G + C% distribution of gene ORF sequence; C, When predicting the start site of a gene ORF sequence, \"edge\" indicative of the anticipated gene’s start codon as uncertain; D, When predicting the gene’s ORF sequence integrity, \"10\" expressive of only the stop codon, \"01\" expressive of only the start codon, \"11\" expressive of neither the stop codon nor the start codon, and \"00\" expressive of the whole gene with both the stop and start codons.")
Sets of Non-redundant Gene Sequences; A, Predicting the length distribution of gene open reading frame (ORF) sequence; B, Predicting the G + C% distribution of gene ORF sequence; C, When predicting the start site of a gene ORF sequence, "edge" indicative of the anticipated gene’s start codon as uncertain; D, When predicting the gene’s ORF sequence integrity, "10" expressive of only the stop codon, "01" expressive of only the start codon, "11" expressive of neither the stop codon nor the start codon, and "00" expressive of the whole gene with both the stop and start codons.
4.2. Differences in Intestinal Flora Between Mild Thalassemia and Healthy Pregnant Women
The relative abundance of Desulfovibrionales in pregnant women with mild thalassemia decreased significantly, compared to that of healthy pregnant women (healthy: 1.48%; mild thalassemia: 0.05%; P = 0.017) (Figure 2A).
 at family level between mild thalassemia and healthy pregnant women; C, Difference in LDA at genus level between mild thalassemia and healthy pregnant women; D, Evolutionary analysis of intestinal flora in mild thalassemia and healthy pregnant women, circles extending from inside to outside representative of classification level from the phylum to the genus, yellow-green nodes indicative of no significant differences in bacterial colonies across groups; yellow areas indicative of a higher abundance of differential colonies in mild thalassemia pregnant women, in contrast to blue areas indicative of a higher abundance of differential colonies in healthy pregnant women.")
Analysis of difference between mild thalassemia and healthy pregnant women; A, Average abundance difference at order level between mild thalassemia and healthy pregnant women; B, Difference in linear discriminant analysis (LDA) at family level between mild thalassemia and healthy pregnant women; C, Difference in LDA at genus level between mild thalassemia and healthy pregnant women; D, Evolutionary analysis of intestinal flora in mild thalassemia and healthy pregnant women, circles extending from inside to outside representative of classification level from the phylum to the genus, yellow-green nodes indicative of no significant differences in bacterial colonies across groups; yellow areas indicative of a higher abundance of differential colonies in mild thalassemia pregnant women, in contrast to blue areas indicative of a higher abundance of differential colonies in healthy pregnant women.
Linear discriminant analysis (LDA) was used to compare the relative abundance of family, genus, and species in mild thalassemia and healthy pregnant women and identify the characteristic intestinal flora of pregnant women with mild thalassemia. The relative abundance of Micrococcaceae and Corynebacteriaceae in the mild thalassemia group significantly increased (P = 0.027 and P = 0.041), compared to that of the healthy pregnant women group; however, the relative abundance of Desulfovibrionaceae and Erythrobacteraceae decreased significantly (P = 0.017 and P = 0.036). In the mild thalassemia group, the relative abundance of Bilophila decreased significantly (P = 0.027), compared to that of the healthy pregnant women group; nevertheless, the relative abundance of Prevotella increased significantly (P = 0.011) (Figures 2B and 2C). While comparing the mild thalassemia pregnant group to the healthy pregnant group, Prevotella stercorea rose (P=0.008), Bacteroides fragilis, a B. species, Roseburia inulinivorans, and a Roseburia species had a reduced relative abundance (P = 0.011) (Figure 3A).

Significant variations in species levels between group of pregnant women with mild thalassemia and group of healthy pregnant women; A, Between group of pregnant women with mild thalassemia and group of healthy pregnant women; B, Between group of pregnant women with mild alpha-thalassemia and group of healthy pregnant women; C, Between group of pregnant women with mild beta-thalassemia and group of healthy pregnant women.
According to the species difference analysis, pregnant women with mild thalassemia belonged to Micrococcaceae at the family level (Figure 2D). Healthy pregnant women belonged to Deltaproteobacteria at the class level, Desulfovibrionales at the order level, and Erythrobacteraceae and Desulfovibrionaceae at the family level; the latter’s results were compatible with those of LDA difference analysis.
4.3. Differences in Intestinal Flora Between Pregnant Women with Mild α-Thalassemia and Healthy Pregnant Women
The relative abundance of Desulfovibrionaceae was significantly lower (P = 0.043) and Mycoplasmataceae significantly higher (P = 0.047) in pregnant women with mild α-thalassemia, compared to that of healthy pregnant women. At the genus level, the relative abundance of Bilophila was significantly lower (P = 0.043) in mild α-thalassemia pregnant women, compared to that of healthy pregnant women; however, the relative abundance of Aerococcus was significantly higher (P = 0.047), and Prevotella, Slackia, Shigella, and Escherichia were also significantly increased (P = 0.043). At the species level, the relative abundance of Escherichia coli in mild α-thalassemia pregnant women increased remarkably (P = 0.043), compared to that of healthy pregnant women; however, the relative abundance of Clostridium saccharogumia and Bacteroides nordii decreased markedly (P = 0.021) (Figure 3B).
4.4. Differences in Intestinal Flora Between Pregnant Women with Mild β-Thalassemia and Healthy Pregnant Women
The relative abundance of Veillonellaceae and Desulfovibrionaceae was significantly lower in mild β-thalassemia pregnant women, compared to that of healthy pregnant women (P = 0.043). However, the relative abundance of Micrococcaceae, Peptococcaceae, and Sphingobacteriaceae increased remarkably (P = 0.021; P = 0.043; P = 0.047). At the genus level, the relative abundance of Tepidibacter was significantly lower in mild β-thalassemia pregnant women than healthy pregnant women (P = 0.047); nevertheless, the relative abundance of Kocuria and Prevotella increased significantly (P = 0.018 and P = 0.021, respectively). The relative abundance of Eubacterium eligens and Bilophila wadsworthia in mild β-thalassemia pregnant women decreased significantly, compared to that of healthy pregnant women at the species level (P = 0.021 and P = 0.043, respectively) (Figure 3C).
5. Discussion
Microorganisms colonize the digestive system and create a natural symbiotic flora or microflora, which is essential for decomposing nutrients and helps resist the settlement of potential pathogens. At present, it is considered that the changes in intestinal flora composition are related to the onset and progression of illnesses, and there are clear disparities between patients with various diseases and healthy individuals (13). In a healthy human gut, the flora will synergize or antagonize one another to preserve intestinal flora’s diversity, stability, and balance, which is crucial for maintaining host health (14). The intestinal flora utilizes specific enzymes and metabolic pathways to degrade nutrients that the body cannot digest or absorb, promoting nutrient absorption and sustaining gut mucosal immunity and systemic autoimmunity (15, 16).
The fetus is generally sterile during the development in the mother’s uterus; therefore, the unborn fetus’s gut is sterile. However, the mother transfers her flora to the newborn neonate during the delivery process. As a result, the mother provides the bulk of the intestinal flora in the early stages of the fetus, protecting the child’s initial and even future health (17). It means that various maternal factors and vertical intestinal flora transfer from the mother will obviously impact fetal development, delivery, and postnatal neonate health. Returning to the present study, the diversity and composition of the intestinal flora in pregnant women with mild thalassemia varied significantly from those in healthy pregnant women, especially at the genus and species levels, representing more profound alterations in intestinal microecology.
Prevotella is a Gram-negative obligate anaerobe that has been linked to insulin resistance, rheumatoid arthritis, diarrheal irritable bowel syndrome, and hyperlipidemia (18, 19) with a similar growth-promoting impact on primary and metastatic colorectal cancer tumors (20). Numerous studies have shown Prevotella to be practically widespread in non-westernized communities that eat a plant-rich diet and less prevalent in European and North American individuals (21-23). Several studies in the recent decade have linked Prevotella to disease-causing qualities, owing to their prevalence in inflammatory situations (24). According to detailed microbial community characterization and metabolome research, Prevotella has a remarkable potential to disrupt the gut microbiome (25) and reduce the amounts of short-chain fatty acids (26). On the other hand, another study proposed that Prevotella might be pro-inflammatory in the stomach by lowering the protective mucus layer (27). Therefore, Prevotella might play an important role in the occurrence and progression of inflammation in pregnant women with mild thalassemia.
Bacteroides fragilis is a Gram-negative anaerobic bacteria that dwell on numerous animals’ lower digestive tracts (i.e., mucosal surface) (28). Bacteroides fragilis colonization has been demonstrated to dramatically enhance the inhibitory action of inflammation-related molecules and the generation of anti-inflammatory cytokines (29). Bacteroides fragilis also possesses immunomodulatory characteristics and is crucial for the development of the human immune system. Intestinal colonization and immune regulation in humans are dynamic. Intestinal colonization and immune regulation in humans are dynamic. The neonate’s initial intestinal flora is primarily derived from the mother’s delivery process and postnatal exposure (30, 31), and B. fragilis, which might be transferred from mother to neonate, is the most abundant bacterium in the newborn’s early digestive tract. The shift in intestinal abundance corresponds to the development of the adaptive immune system (32).
Roseburia inulinivorans in the Firmicutes can prevent colitis by binding to G-protein coupled receptors (33), indicating that B. fragilis and R. inulinivorans have certain anti-inflammatory properties. These two bacteria are much lower in pregnant women with mild thalassemia than healthy pregnant women, rendering them more prone to inflammation. The inflammatory response has long been linked to threatened preterm delivery and birth (34). Simultaneously, the reduction in B. fragilis might affect the development of the fetal immune system.
Escherichia coli is the most prevalent facultative anaerobic bacterium in the human gut flora and one of the main opportunistic pathogens of nosocomial infection (35). It can cause body inflammation via immune responses, resulting in diarrhea, peritonitis, cystitis, and cholecystitis (36). Colon cancer-related anemia could increase the relative abundances of E. coli as one of the intestinal flora imbalance-inducing bacteria (37). Clostridium can be classified into two main types, namely beneficial and harmful. Clostridium saccharogumia is an intestine probiotic Clostridium that can convert Secoisolariciresinol diglucoside into lignan Enterolactone and Enterodiol, which could prevent breast cancer, colon cancer, atherosclerosis, and diabetes (38, 39).
Bacteroides nordii can oxidize primary bile acids to secondary bile acids, protect intestinal epithelial cells, and fight infections, modulating bodily health and intestinal flora composition (40). In comparison to healthy pregnant women, mild α-thalassemia pregnant women are prone to intestinal microecological imbalance, bile acid metabolism disorders, and increased plasma bile acids, leading to adverse consequences, such as premature delivery and respiratory distress or death.
Eubacterium is a symbiotic anaerobic bacterium found in the human intestine that is part of the dominant flora. Eubacterium can effectively reduce blood glucose levels, protect intestinal epithelial barriers, and treat inflammatory bowel disease (41). Bilophila wadsworthia is an obligatory anaerobic Gram-negative bacillus with high bile salt tolerance in the Desulfovibrionaceae family. Bile acids can promote the proliferation of B. wadsworthia in the intestine. The main treatment for thalassemia is blood transfusion coupled with chelation therapy with iron remover. The major treatment approach for β-thalassemia is blood transfusion chelation therapy coupled with an iron removal agent. Regardless of whether the patient undergoes blood transfusion treatment, the iron in the body will be overloaded, with most of it being stored in the liver (42).
Excess iron has been shown to harm the liver and impair bile acid production (43). The relative abundance of B. wadsworthia in the intestines of mild β-thalassemia pregnant women reduced, which might be attributed to reduced bile acid production induced by iron overload. Bile acids act as a detergent in lipid metabolism and are essential for metabolism and immunological regulation. They can maintain a steady state of intestinal flora, improve mucosal barrier defense, and inhibit bacteria growth. Reduced bile acid synthesis and damage to the intestinal epithelial barrier might result in intestinal bacterial translocation, activation of the inflammatory immune defense mechanism in pregnant women with mild thalassemia, stimulation of the systemic inflammatory response, and threatened premature birth or a negative impact on the newborn’s health.
However, there are still numerous areas to be improved in the present study. The most serious issue is the difficulty of collecting samples, which leads to a small number of samples in this study. The selected subjects in this study only represented the composition of the intestinal flora in local samples, and the subjects’ factors affected the results. Due to the limitation of the existing mNGS methodology, numerous strains have not been discovered. As a result, the investigation of flora still meets hindrance everywhere. In addition, the intestinal flora is dynamic, and this study can only show the state of the research object at a certain point in time. Therefore, long-term practical dynamic observation should be made in the follow-up, which might be more valuable for research.
5.1. Conclusions
In conclusion, the present study results showed that the diversity and composition of the intestinal flora in pregnant women with mild thalassemia vary significantly from those in healthy pregnant women, especially at the genus and species levels, representing more profound alterations in intestinal microecology. The disorder of the intestinal flora has a certain negative impact on the health of pregnant women with mild thalassemia, the fetus, and the process of pregnancy and childbirth.
