Prostate cancer (PCa) is the most diagnosed cancer in adult men and the second cause of cancer deaths in North American and European men. It is estimated that one of the six men may be afflicted with PCa during their lives (
1). There are such handful factors increasing the risk of PCa as: age, race, genetics, and lifestyle (
2,
3,
12–
21,
4,
22–
24,
5–
11). The routine screening tests for diagnosis of PCa are prostate specific antigen (PSA) blood test and digital rectal examination (DRE)
25-26. In cases with high PSA level or any abnormal observations in DRE, prostate biopsy serves as an invasive method. CT-Scan, MRI, and prostate scintigraphy are main imaging techniques for detection of metastasis (
27). There are some known biomarkers associated with PCa like (
28) prostate stem cell antigen (PSCA) (
29–
31), prostate specific G-protein coupled receptors (PSGR), PSA (
32–
37) and PSMA (
32–
35). Of these markers, PSMA has gained more interest so as to early detection of PCa due to several reasons: 1) Over-expression on prostate cancer cells as membrane antigens 2) Broadly expression at all stages of prostate tumors 3) Up-regulation in androgen-insensitive and metastatic tumors 4) The absence on normal prostate cell surface with restricted expression in the brain, kidney, and small intestine. 5) Intrinsic enzymatic activity which makes it possible to design ligand-targeted drugs 6) Receptor-mediated Endocytosis of PSMA-targeted ligands leads to ligand retention into tumor cells.
Two distinct types of probes are used for targeting PSMA. The first groups were designed based on monoclonal antibodies (MAbs) which are not widely applied today (
38). Low molecular weight pepidomimetics as PSMA inhibitors are the second attractive agents because of easy synthesis, high affinity, better tissue penetration, rapid clearance, and low immunogenicity (
39). According to published studies, the active site of PSMA is of a funnel-like structure with an arginine patch that can be occupied by two main parts of PSMA inhibitors: glutamic acid or derivatives which ensures high affinity binding and zinc binding groups such as: Phosphonate, Sulfamide, Hydroxamate, Carbamate, and Urea moiety (
40–
42).
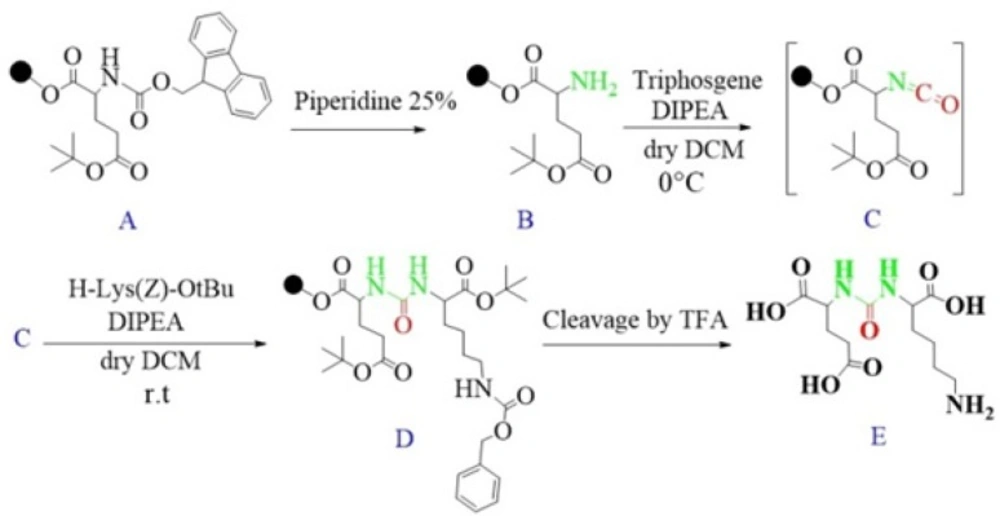
Synthesis of glu-urea-lys (EUK) in solution phase (45).
Synthesis of peptide containing glu-urea-glu (DUPA) motif for PSMA targeting. At first DUPA was formed by aqueous reaction between L-Glutamic acid di-tert-butyl ester and triphosgene. Then, it was conjugated to N-terminus of peptide residue. This method can’t be used for attaching the other pharmacophor of PSMA due to their absence of free Carboxylate (47).
Liquid phase Synthesis of tyr-urea-glu (TUG) as a dipeptide PSMA inhibitor (50).
Synthesis of isocyanate intermediate under solution phase and then coupling with resin immobilized (2-chloro-tritylresin) ε-allyloxycarbonyl (Alloc) protected lysine in order to prepare glu-urea-lys (EUK)-containing peptides. During this method, there was no simple way to confirm the formation of isocyanate in solution phase (46).
Schematic illustration of the synthesis route of glu-urea-lys (EUK) as a PSMA inhibitor.
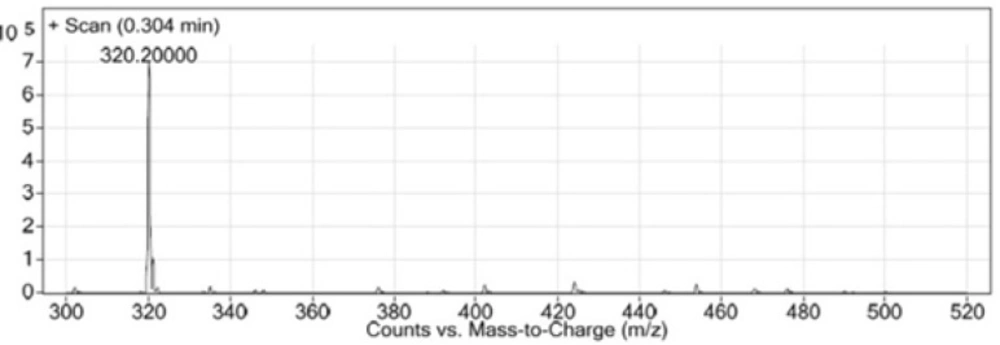
LC-MS chromatogram of synthesized Glu-Urea-Lys (EUK).
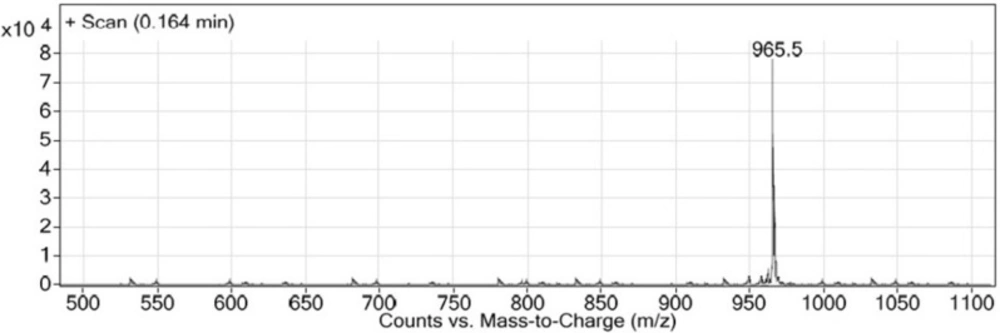
LC-MS chromatogram of Glu-Urea-Lys (OMe)-GABA-Tyr-Tyr-GABA-HYNIC.
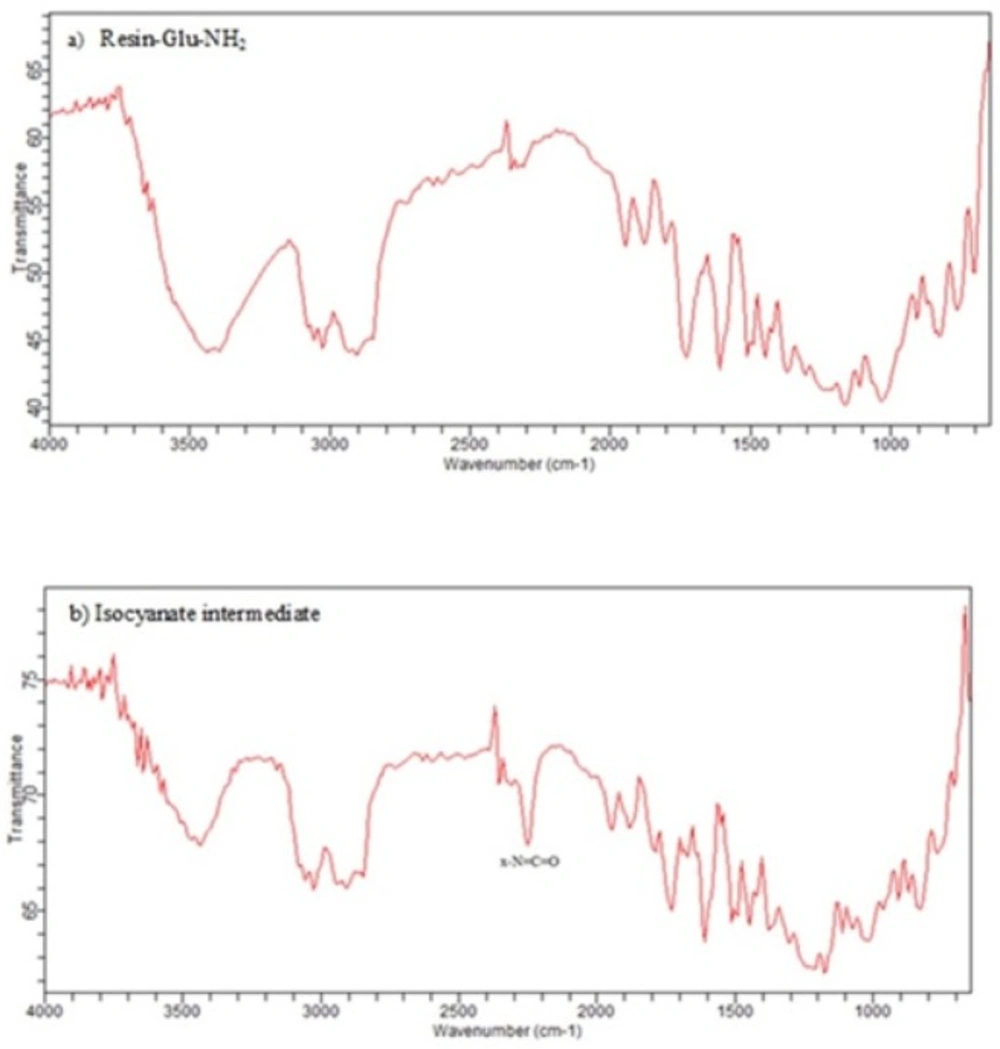
IR spectra of a) Deprotected glutamate bound to wang resin b) The corresponding isocyanate intermediate on wang resin. X-N=C=O stretch is seen in the 2270 cm–1 region of the IR (b) while it is absent in IR spectrum of resin-Glu-NH2 (a).
The basic chemical structure of most PSMA inhibitors which are now in pre-clinical and clinical studies is urea-based peptide. The pharmacophore, Glu-urea-R, specifically binds to PSMA and inhibits its activity. The Glu-Ureido-based inhibitors and its analogs are the most desirable probes since 2001. Relative simple synthesis and biological stability because of planar structure and neutral charge of ureido group might have made it
44-45. Four general urea containing pharmacophores have been introduced so far which are analogue of dipeptides: glutamate-urea-glutamate (EUE), glutamate-urea-cysteine (EUC), glutamate-urea-tyrosine (GUT), and glutamate-urea-lysine (EUK) (
45). Unlike MAbs, peptide scaffolds as small molecules are synthesized easily by SPPS and more resistant in extreme radiolabeling conditions (temperature, pH). This is why they have been compared to the best candidate for designing radiopharmaceuticals (
39).
Synthesis of urea-based PSMA inhibitors includes two steps: 1) isocyanate intermediate formation and 2) urea bond formation (
43,
45–
50). The isocyanate intermediate is synthesized from the reaction of appropriate amino acid with tri/diphosgene in liquid phase under controlled conditions of temperature and pH. In the next step, the urea bond is formed from the reaction of amino acid free amine with isocyanate. Overall, these current methods involve conjugation of free amine whether in liquid phase
44,46,51-52 or bound to solid phase (
46) with isocyanate formed in solution phase before.
In recent years, a variety of radioligands targeting prostate-specific membrane antigen (PSMA) have been clinically developed as a new class of radiopharmaceuticals for prostate cancer. Sigurdsson
et al. in 1999 prepared isocyanate from aliphatic amines using trichloromethyl chloroformate (diphosgene) at 0 °C in presence of the non-nucleophilic base (
51). Kozikowski
et al. in 2001 reported the synthesis of urea-based glutamate carboxypeptidase II (NAALADase) inhibitors in which triphosgene has been used for carbonylation of amine in liquid phase at -78 º C. Afterwards, the corresponding intermediate reacted with free amine of the other amino acid to form urea moiety (
43). Hiller
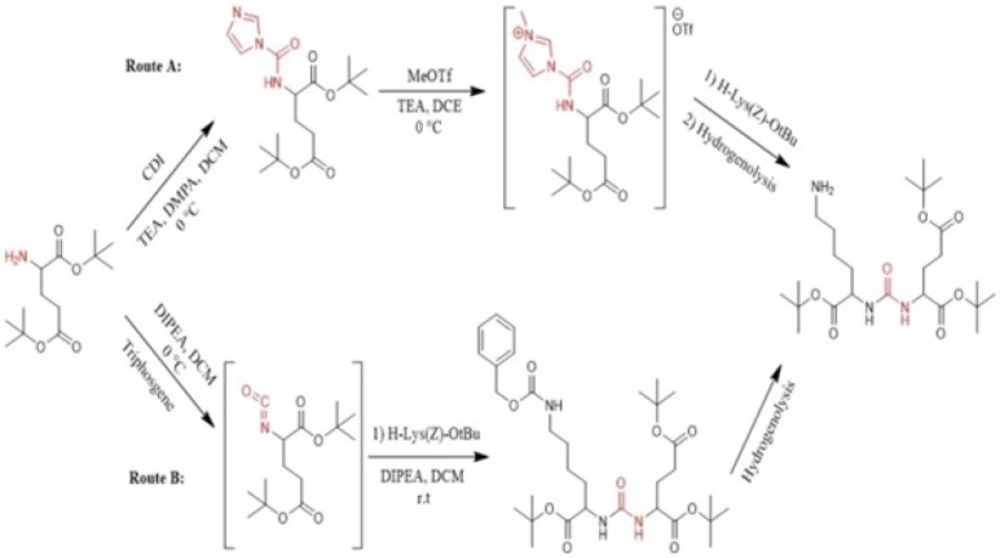
et al. in 2009 synthesized a series of Glu-urea-X heterodimers as PSMA inhibitors, which X is a derivatized lysine (Lys). The compounds were radioiodinated and theirs affinity to prostate cancer cells were determined. The urea linkage was synthesized in liquid phase utilizing two routes: acylimidazole intermediate afforded by Carbonyldiimidazole (CDI) (route A) and isocyanate intermediate prepared by Triphosgene (route B) (
Figure 1) (
45).
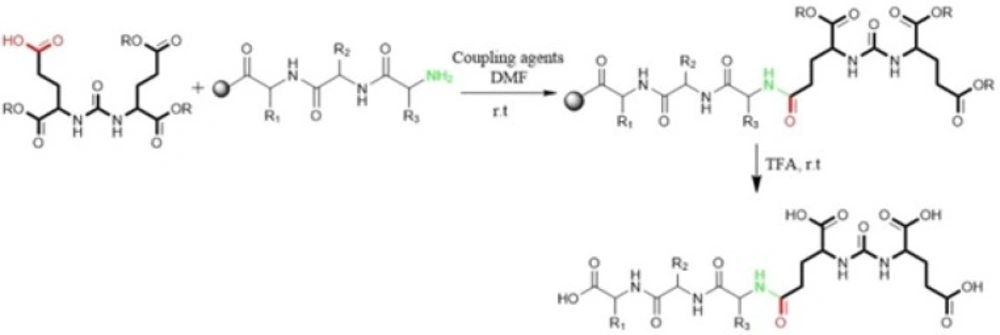
Kularatne
et al in 2009 prepared a series of (Glu-urea-Glu)-based PSMA inhibitors. At first, The Glu-urea-Glu pharmacophore (EUE) was synthesized through isocyanate intermediate formation in liquid phase using triphosgene under controlled conditions. Then, one of the carboxylate groups in the Glu-urea-Glu was de-protected via catalytic hydrogenation and subsequently conjugated to N-terminal of resin-bound peptide to furnish final peptide. Actually, in this procedure both solid phase and solution phase peptide synthesis techniques have been applied (
Figure 2) (
47).
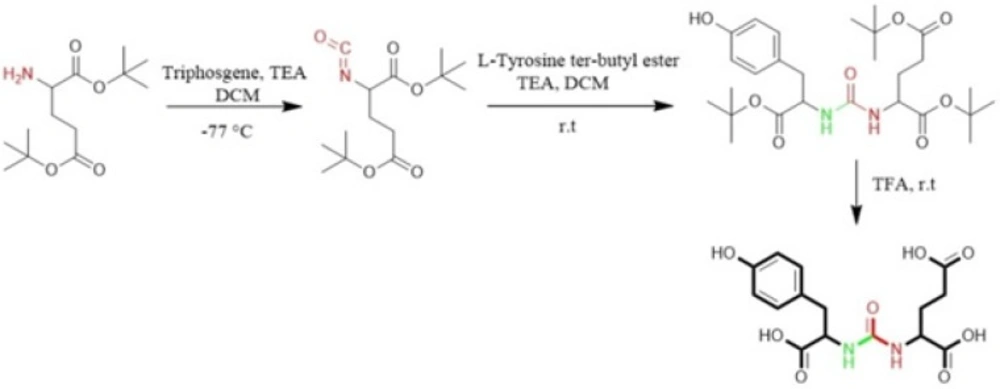
Al-momani
et al. in 2012 only took advantage of solution phase peptide synthesis method to prepare Glu-Urea-Tyr. The reaction of triphosgene in CH
2Cl
2 with bis (tert-butyl)-L-glutamate.HCl using triethylamine as a base, at -77 °C under argon, resulted in isocyanate intermediate. In next step, L-Tyrosine tert-butyl ester were activated by triethyamine and conjugated with isocyanate to afford urea linkage (
Figure 3) (
50).
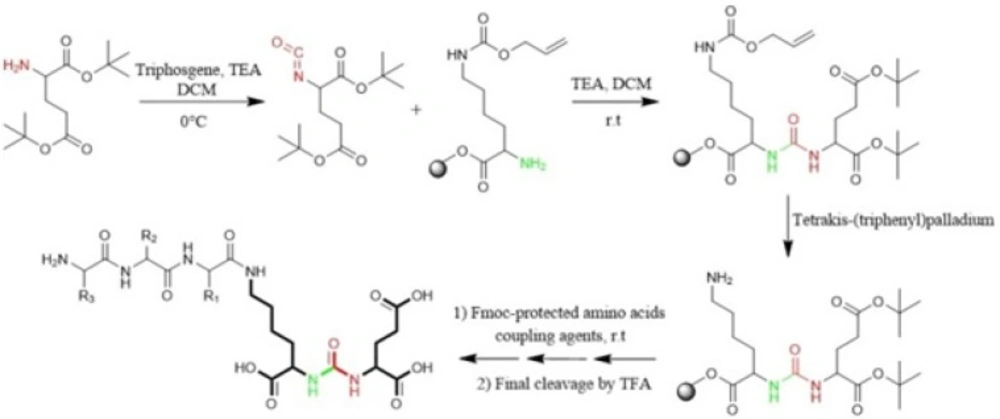
Eder
et al in 2012 synthesized Glu-NH-CO-NH-Lys (Ahx)-HBED-CC. To form urea bond, the isocyanate of glutamyl moiety (Glu-N = C = O) was synthesized in liquid phase and then reacted with free α-amino group of resin-bound ε-allyloxycarbonyl (Alloc) protected lysine. So, the peptide chain elongation was completed in solid phase (
Figure. 4) (
46).
Zhang
et al. in 2016 used similar liquid phase procedures using triphosgene under controlled conditions (temperature, pH, anhydrous and N
2) for synthesis of four analogs of Glu-Urea-R. They also determined the affinities of synthesized compounds to PSMA (
49).
In the present study, we introduce a rapid method for solid phase synthesis of isocyanate and urea moiety using peptide coupling reagents.