Materials
General molecular biology reagents including RNX-Plus, Tris-Base, SDS (Sodium dodecyl sulfate), Acrylamide, Bisacrylamide and Agarose were purchased from CinnaGen, Iran. Most of the required enzymes including pfu DNA polymerase, MMLV-rt(Moloney Murine Leukemia Virus reverse transcriptase), T4 DNA Ligase, EcoRV, BamHI, SacI and BglII were obtained from Fermentase. 35S-CaMV plasmid and pGreen II 0179 were purchased from John Innes Centre, UK. Agrobacterium tumefaciens LBA 4404 and Nicotiana tabacum cell line were kindly donated by NIGEB (National Institute of Genetic Engineering and Biotechnology) and Dr Ghanati (Tarbiat Modares University), respectively. All requirements for plant cell culture media including major and minor minerals; hormones (such as Indole acetic acid, Naphthyl acetic acid, Kinetin, Acetosyringon), vitamins and sugars (Such as Myoinositol and Sucrose) were provided from Merck, Germany. Antibiotics including Ampicillin, Kanamycin, Tetracycline, Hygromycin, Streptomycin and Cefotaxime were purchased from Sigma. Phenyl Methyl Sulfonyl Fluoride (PMSF) protease inhibitor and nitrocellulose membrane were acquired from Roche and Protino Ni-TED (Nickel-Tris(carboxymethyl)ethylene diamine) packed chromatography column was purchased from Machehery-Nagel, UK. Bio-Rad detergent compatible Kit and New England BioLab Bovine Serum Albumin were used to determine protein concentration. Recombinant TRAIL (ab168898) as standard along with TRAIL polyclonal antibody (ab2435) and anti-rabbit IgG secondary antibody (ab131365) were purchased from Abcam. A549 cell line (ATCC® CCL-185™) as a cancer model was obtained from Pasteur Institute of Iran.
Methods
Preparation of Soluble Human TRAIL expression gene fragment
In order to obtain soluble human TRAIL gene fragment, at first total RNA of human peripheral white blood cells was extracted using RNX-Plus reagent as manufacture’s instruction manual. Then, cDNA library was constructed by utilizing MMLV- reverse transcriptase and oligo-dT primers. Afterward, to obtain the extracellular region fragment of TRAIL, PCR was carried out on cDNA library by exploiting
pfu DNA polymerase and specific (F1 and R1) TRAIL primers (
Table 1 and
Table 2). Subsequently, nested PCR reaction was performed by F2 and R2 primers (
Table 1 and
Table 2) on obtained fragment to attain encoding region of soluble human TRAIL. The final achieved fragment was confirmed by sequencing technique.
| Primers | Sequences from 5' to 3' | Descriptions |
|---|
| F1 | ACAGCCCCTGCTGGCAAGTC | Specific Forward |
| R1 | TTAGCCAACTAAAAAGGCCCCGA | Specific Reverse |
| F2 | GTGAGAGAAAGAGGTCCTCAGAGAG | Nested Forward |
| R2 | GGTACCGCCAACTAAAAAGGCCCCA | Nested Reverse |
| F3 | GGGATCCAACAATGGTGAGAGAAAGAGGTCCTCAGAGAG | BamHI + Kozak |
| R3 | ATGATGACCTCTGCCAACTAAAAAAGCCCC | His-Tag |
| R4 | ATGATGATGATGATGATGACCTCTGCCAAC | His-Tag |
| R5 | GGAGCTCTTAATGATGATGATGATGATGACCTCTGCCAAC | His-Tag + SacI |
| PCR primers | Initial Denaturation | (PCR Cycles)*30 | Final Extension |
|---|
| F1 R1 | 95 oC (5min) | 95 oC (45 Sec)61 oC (40 Sec)72 oC (1 min) | 72 oC (10min) |
| F2 R2 | 95 oC (5min) | 95 oC (40 Sec)60 oC (40 Sec)72oC (1 min) | 72 oC (10min) |
| F3 R3 | 95 oC (5min) | 95oC (40 Sec)65 oC (40 Sec)72oC (1 min) | 72 oC (10min) |
| F3 R4 | 95 oC (5min) | 95oC (40 Sec)63 oC (40 Sec)72oC (1 min) | 72 oC (10min) |
| F3 R5 | 95 oC (5min) | 95oC (40 Sec)66 oC (40 Sec)72 oC (1 min) | 72 oC (10min) |
Providing expression and translation controlling regions
With the aim of expressing soluble human TRAIL in plant cell system, at first, “ plant compatible 5ʹ translation initiator Kozak sequence” and “C-terminal 6 His-Tag purification facilitator” fragments were added to TRAIL encoding gene using sequential PCRs via respective (F3) and (R3, R4, R5) primers (
Table 1 and
Table 2). All PCR reactions were carried out by
pfu DNA polymerase and the accuracy of the reactions was confirmed by sequencing technique. The obtained cassette was cloned between the
BamHI and
SacI regions of 35S-CaMV plasmid possessing plant specific promoter and terminator regions (
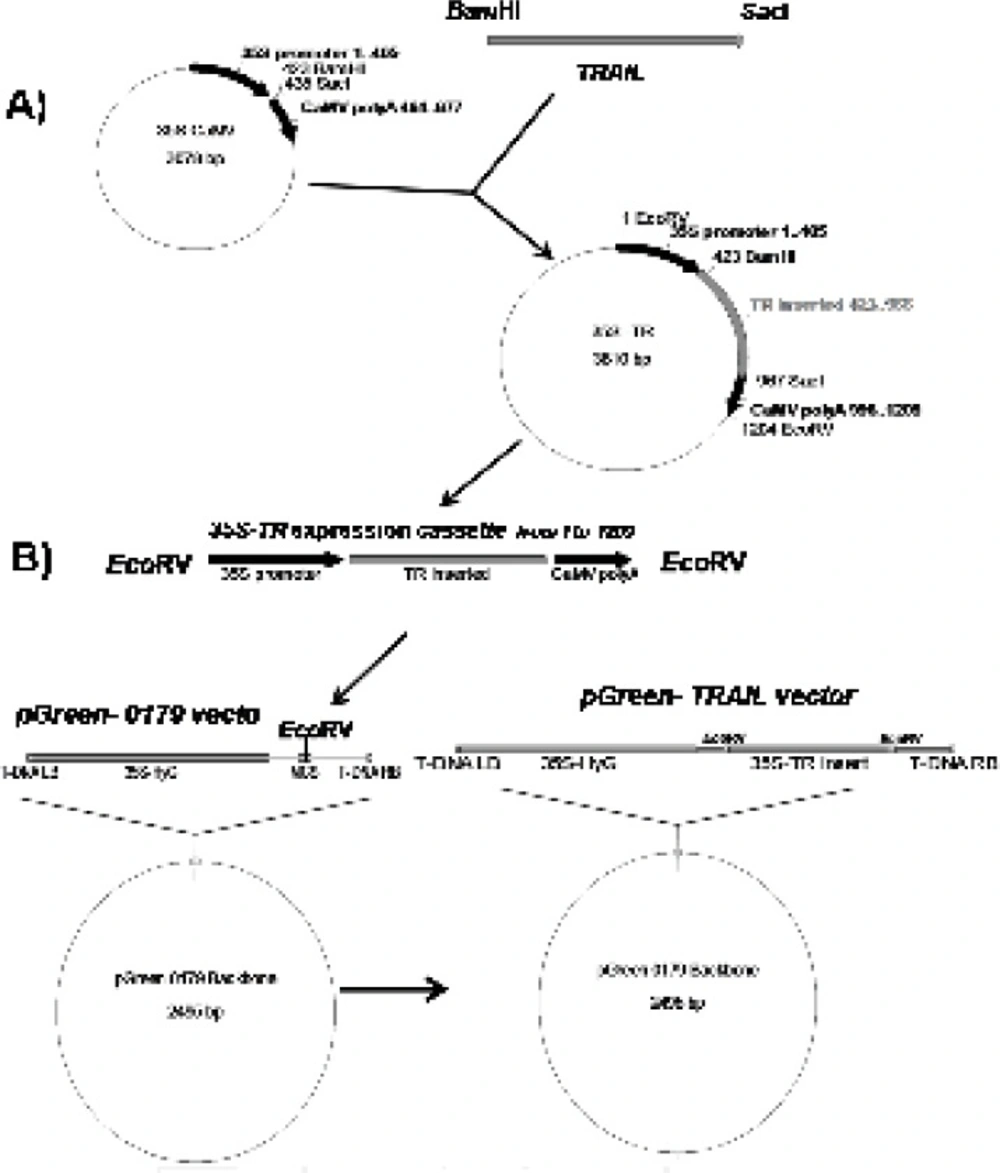
Figure 1A). Finally, the designed expression construct was sub-cloned into the
EcoRV region of pGreen II 0179 plant expression vector mainly to provide both selectable markers and
A. tumefaciens transferable oncogenic DNA (T-DNA) regions (
Figure 1B).
Schematic diagram of soluble human TRAIL cloning into intermediate 35S CaMV vector and pGreen-0179 plant expression vector. Panel A) represents cloning of TRAIL into BamHI and SacI regions of 35S-CaMV plasmid to obtain 35S promoter-TR inserted-CaMV polyA (1200 bp) fragment. Panel B) represents cloning of 35S promoter-TR inserted-CaMV polyA fragment into EcoRV region of the pGreen-0179 T-DNA to obtain final pGreen-TRAIL vector. LB and RB (Left border/right border) stands for left border and right border respectively.
Cloning into Agrobacterium tumefaciens
A.tumefaciens LBA4404 was grown on broth Luria-Bertani medium included streptomycin (100 μg/mL) at 25
oC, 200 rpm for 48 hours. The final obtained TRAIL expressing pGreen vector and replication facilitator pSoup helper plasmid (
27) were co-transformed to
A. tumefaciens LBA 4404 using previously described freeze-thaw method (
28). Transformed bacteria were selected on Kanamycin (50 μg/mL), Tetracycline (2 μg/mL) and Streptomycin (100 μg/mL) selection medium. To induce the virulent genes of
Agrobacterium, the transformed bacteria were grown on broth selection medium containing 100 μM Acetosyringon for at least 2 hours.
Transformation and selection of Nicotiana tabacum cells
N. tabacum callus cells were grown at 25
oC in the darkness condition on solid LS medium (Linsmaier & Skoog Medium) (
29) supplemented with Indole acetic acid 3 μg/mL, Naphthyl acetic acid 3 μg/mL, Kinetin 100 ng/mL, Myoinositol 100 μg/mL and Sucrose 30 mg/mL Since Zn
2+ plays important role in proper assembly of the TRAIL, two fold of Zn
2+ ion regular concentration was used for LS medium preparation. Transformation of
N. tabacum cells was performed by co-cultivation technique (
30) with some modifications. At first, 10 mL of freshly activated transformed
A. tumefaciens (OD: 1, 600 nm) (Optical Density) was centrifuged (3000 rpm, 5 min, 25
oC) and the medium was replenished by 5 mL modified LS medium containing 100 μM Acetosyringon. Afterward, one gram of freshly grown
N. tabacum cells was added to the bacterial culture and was incubated at 100 rpm about 2 h at 25
oC. The incubated cells were centrifuged (1000 rpm, 10 min, 25
oC) and the cells pellet was placed on the sterile filter paper for 5 min to wipe excess water. Inoculated plant cells were subsequently transferred to the solid modified LS medium containing 100 μM Acetosyringon and were kept for two days at 25
oC in the darkness condition. Remained
Agrobacterium cells were eradicated by means of sub-culturing on medium containing Cefotaxime (200 μg/mL). Finally, transformed
N. tabacum cells were screened through frequent sub-culturing on selection medium containing Hygromycin (30 μg/mL) and Cefotaxime (200 μg/mL) every two weeks. Selected
N. Tabacum cells in the week 8
th were used for the rest of the experiments.
Total protein extraction and TRAIL purification
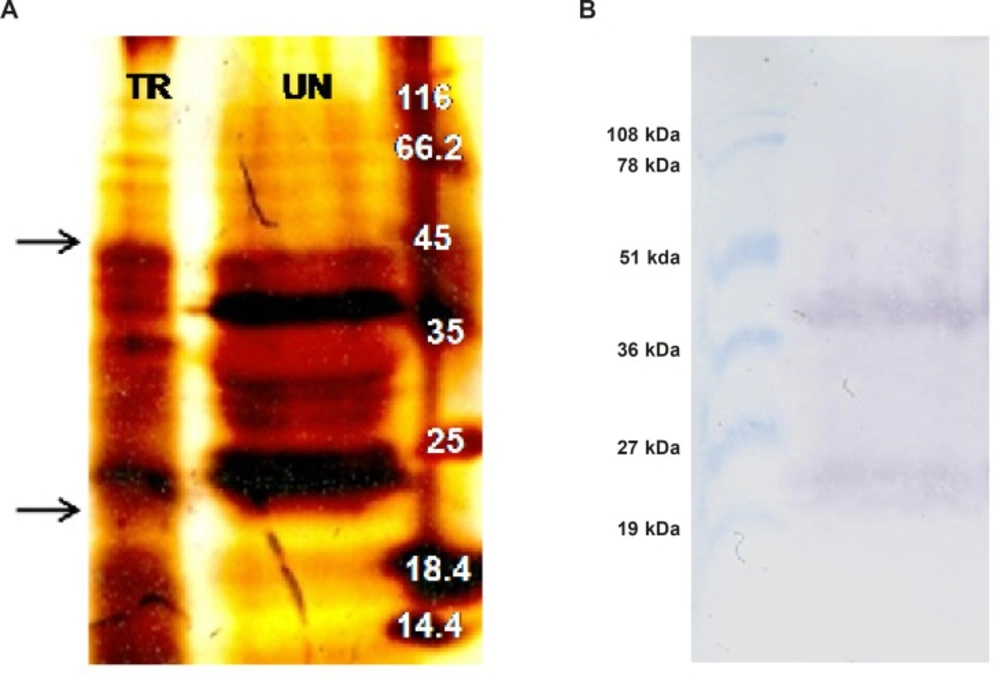
Two grams of transformed N. tabacum cells were suspended into 5 mL ice cold non-reducing extraction buffer (PBS pH: 7.4, Glycerol 10%, PMSF 1 mM) (Phosphate Buffered Saline) and were homogenized using Heidolph silent crusher (20000 rpm, 45 seconds). The mixture was placed on ice for 10 min and was centrifuged twice (12000 rpm, 15 min, 4 oC). The supernatant was filtrated through 0.45 μm filter to remove remained solid materials. Meanwhile, total protein concentration was measured using Bio-Rad DC kit (based on Lowry method), where bovine serum albumin was utilized as a standard. To purify recombinant TRAIL, total protein extract of transformed N. tabacum cells was passed through pre-equilibrated Ni-TED pre-packed column and the purity of the TRAIL was confirmed by SDS-PAGE.
Western blot analysis
A total of 1 mg of acetone precipitated total protein extract from both transformed and untransformed plant cells was denatured by boiling in 100 μL non-reducing loading buffer (0.225 M Tris Cl, pH 6.8, 50% glycerol, 5% SDS) for 5 min. Subsequently, 200 μg of the protein samples along with pre-stained protein marker (Biobasic, RM0011, Korea) were separated on a 15% SDS-PAGE. Afterward, the separated proteins were transferred to a nitrocellulose membrane by semi-dry technique (Towbin buffer, 200 mA, 2 h, Apelex PS 304). The proper transfer of proteins to the membrane was indicated by the color of the pre-stained protein marker which remains the same even after blotting. After attaching the proteins to the nitrocellulose membrane by ultra violet cross linking method (120 m Joules, 1min, UviTech), the membrane was blocked by incubating overnight in PBS buffer (pH 7.4) containing 5% (w/v) skimmed milk at 4 oC. Following washing three times in Tris-Buffered Saline (TBS: 20 mM Tris-HCl, 140 mM NaCl, pH 7.5) at 200 rpm for 10 min, the membrane was incubated for 2 h (80 rpm) with a rabbit polyclonal antibody of TRAIL (1:1000; Abcam, ab2435) in TBS-T buffer (TBS plus 0.1% Tween-20, v/v) containing 1% (w/v) skimmed milk. Afterward, the membrane was subjected to three times washing procedures with TBS and TBS-T buffers (10 min, 200 rpm). Subsequently, the membrane was incubated for 1 h (80 rpm) with alkaline phosphatase conjugated anti-rabbit IgG secondary antibody (1: 5000; Abcam, ab131365) in TBS-T buffer, containing 1% (w/v) skimmed milk. After several washing steps (5 min, 200 rpm) with TBS and TBS-T buffers, the membrane was incubated with Bromo Chloro Indolyl Phosphate (BCIP) and Nitro Blue Tetrazolium (NBT) reagents in alkaline phosphatase buffer (100 mM NaCl, 5 mM MgCl2, pH 9.5) for color development. Finally, the color development was stopped by washing the membrane with distilled water.
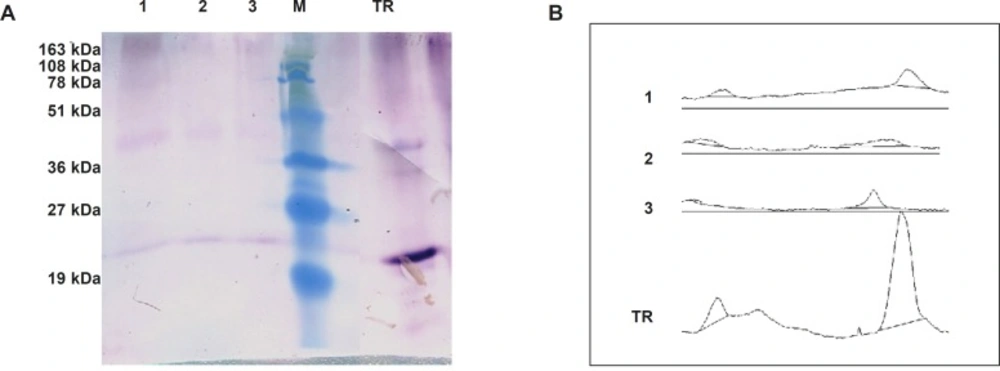
Semi-quantitative western blot analysis
A total of 200 μg from crude protein extract of recombinant N. tabacum samples and 1 μg of recombinant human TRAIL (ab168898) as a standard were exploited to SDS-PAGE and semi quantitative western blot analyses. The intensity of developed bands was analyzed using ImageJ software and data were utilized to calculate approximate amount of TRAIL production in transformants.
Functional assay
The anti-proliferation activity of TRAIL on A549 cells line was determined using MTT assay as previously described (
31). Briefly, A549 cells were cultured at a seeding density of 4.0 × 10
4/cm
2 in 96-well micro plate, each containing 200 μL of the growth medium (RPMI-1640). The cells were grown in the humidified incubator at 37
oC with 5% CO
2, until they reach about 40% confluence. Total protein extracts of both transformed and untransformed
N. tabacum cells were concentrated with Centricon filter concentrators (10 kDa cutoff). A total of 200 μg of concentrated protein extracts, as well as 1 μg His tag purified TRAIL were added to each well, following by further incubating for 48 h at cultivation condition. Correspondingly, 100 ng recombinant human TRAIL (ab168898) and 50 μL extraction buffer were used as positive and negative controls, respectively. After incubation period, the medium was replaced with 200 μL fresh medium containing extra 50 μL of MTT solution (2 mg/mL in PBS), and the cells were incubated for an additional 4 h at 37
oC. Afterward, the medium was completely removed and 200 μL DMSO (Dimethyl sulfoxide) in addition to 25 μL Sorenson buffer (0.1 M glycine, 0.1 M NaCl, pH 10.5) were added to each well. The absorbance of each well was measured by employing a BioTek Synergy 3 micro plate reader at 570 nm. Obtained data were demonstrated as Mean ± SD. Results were implemented by Excel (version 2007) and SPSS (version 16). Statistical analyses between mean values were performed using one-way analysis of variance (ANOVA) and post test of least significance difference (LSD). p-value less than 0.05 was considered as significant difference.