1. Introduction
The human tail is an extremely rare congenital anomaly that typically manifests at birth or in early childhood. Tojima and Yamada., who reviewed case reports of human tails in English, French, Japanese, Italian, and German literature in 2020, reported only 195 cases of human tails as of 2017 (1). The birth of a baby with a tail can significantly impact the parents' psychological well-being and may lead to social and cultural stigma for the baby and the family, potentially resulting in delayed diagnosis and treatment, even into young adulthood (2).
Various classifications exist for human caudal appendages, but the oldest and most widely used was described by Dao and Netsky in 1984. They categorized these tails into two main groups: True tails and pseudo-tails. True tails contain adipose, connective, and muscle tissue and lack any bony structures, while pseudo-tails are superficial manifestations of an associated structural anomaly, such as lipomas or vertebral elongation (3). Studies have shown that human tails are associated with other malformations, such as open or occult spinal dysraphism, in approximately 40% of reported cases (2, 4-7).
In 2020, Tojima and Yamada proposed a new classification (1). They categorized human tails into four types based on the content and location of the lesion: Type Ia involves a tail caused by protrusion of the coccyx, Type Ib includes tails with bony or cartilaginous non-coccygeal components, and Types IIa and IIb consist of tails lacking any bony or cartilaginous materials, located above or below the natal cleft, respectively. Although human tails are primarily found in the lumbosacral region, cervical tails have also been reported (8).
Here, we discuss a neonate with a tail-like structure that seems potentially detectable through a mid-gestation anomaly scan, along with the associated genetic findings. The previous pregnancy was also complicated by neural tube defects. To the best of our knowledge, no similar report of soft tissue tails with concomitant whole exome sequencing findings has been documented in the English language to date.
2. Case Presentation
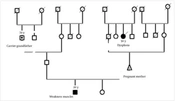
A 3-week-old infant was admitted to Baqiyatallah Hospital (Tehran, Iran) with a congenital elongated midline cutaneous structure resembling a tail in the lumbosacral region. He was the son of consanguineous parents, and his 32-year-old, gravida 2, para 2 mother had experienced an uneventful pregnancy and postpartum course. Notably, her first pregnancy had been diagnosed with a neural tube defect (NTD), most likely a large lumbosacral meningomyelocele, which led to a legal abortion at 18 weeks of gestation in 2020. During the recent pregnancy, serum aneuploidy screening and fetal echocardiography were both within normal limits. The mother had taken 1 mg of folic acid daily throughout her entire pregnancy. She was a housewife and denied any teratogenic exposure during her pregnancy.
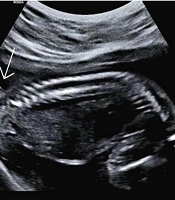
The newborn was delivered vaginally at 40 weeks of gestation in a rural area of Arak province. Unfortunately, due to social stigma, the parents had delayed seeking medical care but eventually came to our center out of extreme concern for their child. Upon retrospective review of the medical records, it was noted that the available anomaly scan images showed a probable cutaneous lesion in the lumbosacral region, in exactly the same location as the postnatal tail. However, the spinal bony structure appeared normal (Figure 1).

Figure 1.
Sonographic imaging of the fetus with a suspected superficial lesion in the lumbosacral region
The infant’s neurological examination revealed no focal deficits in either the lower or upper extremities. The anal sphincter displayed normal tone, and the perineum appeared normal. A 12 cm, skin-covered, soft, and non-tender tail-like lesion was present in the lumbosacral region. The lesion did not contract or move, and no hard components were palpable. Local melanocytosis was also evident in the tail region and upper buttocks. Additionally, the parents reported no significant growth of the lesion since birth (Figure 2).

Figure 2.
Tail in the midline of the lumbosacral region
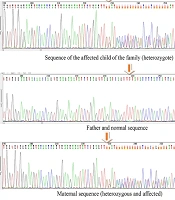
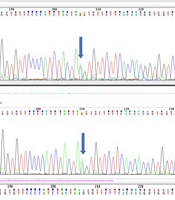
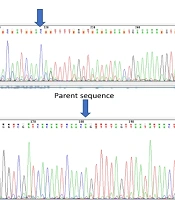
To rule out any underlying spina bifida, lumbosacral spine X-ray imaging was performed, which was unremarkable. Pelvic MRI also ruled out any occult spinal dysraphism. Additionally, abdominopelvic and brain sonographic scans were unremarkable, and neonatal echocardiography revealed no pathological findings. Moreover, since the parents were related and the previous pregnancy had been complicated by an NTD, genetic consultation was performed, followed by whole exome sequencing (WES), the results of which are shown in Table 1.
Table 1.
Findings of the Infant’s Whole Exome Sequencing
| Gene | Protein Alteration | Chromosome Position/No of Changed Nucleotide | Zigosity | Related Phenotype | Inheritance Pattern | Clinical Significance |
|---|---|---|---|---|---|---|
| HOXA13; NM_000522:exon1:c:G196T | p.A665 | 7 (27239501) | Het | Hand-foot-uterus syndrome | AD | VUS |
| LBR; NM_002296:exon8:c.A899G | p.Y300C | 1 (225600341) | Het | Pelger-Huet anomaly with mild skeletal anomalies | AR | VUS |
| DYNC2H1; NM_001080463:exon12:c.C 1774T | p.L592F | 11 (102995941) | Het | Short-rib thorasic dysplasia 1 with or without polydactyly | AR | VUS |
| DNAAF1; NM_001318756:exon5:c.c824t | p.P275 | 16 (84205869) | Het | Ciliary dyskinesia, primary, 13 | AR | VUS |
| GPR161; NM_001267611:exon2:c.T47A | p.L16Q | 1 (47746675) | Het | Pituitary_stalk_i nteruuption_syn drome | AR | VUS |
| variants unrelated to phenotype in WES data | ||||||
| TYR; NM_000372:exon4:Cc1217t | p.P406L | 11 (89017973) | Het | Albinism, oculocutaneous | AR | pathogenic |
Abbreviations: VUS, variant of uncertain significance; AD, autosomal dominant; AR, autosomal recessive; Het, heterozygote; WES, whole exome sequencing.
Surgical excision was planned; however, the parents opted to have the surgery at another hospital due to financial issues related to their insurance coverage and the distance to their home. Approximately one month later, the surgery was performed. According to the operation notes, the surgeon removed the tail through an elliptical incision. The tail had no communication with the spinal canal. A telephone follow-up was conducted to obtain the histopathological diagnosis of the lesion, and microscopic examination revealed only mature adipose tissue. Further telephone follow-up was performed when the child was 2 years old, revealing appropriate neurodevelopmental status.
3. Discussion
Human embryos develop a tail with 10 - 12 caudal vertebrae during the 5th to 6th weeks of gestation. This tail begins to regress, and the fusion of the vertebrae leaves a vestigial coccyx. The tail typically disappears by the end of the 8th week, though the timing may vary. Persistence of the most distal avertebrate remnant of this embryonic tail can lead to a permanent tail (9).
Although cases similar to ours are usually diagnosed at birth or in early childhood, earlier diagnoses, such as those in the first trimester, have been reported. For example, Garange et al. reported a human tail at 13 weeks of gestation, where sonographic follow-ups showed its disappearance by 21 weeks. However, intrauterine fetal death occurred three weeks later, and postmortem autopsy revealed thin limbs, clinodactyly of the fifth finger, partial syndactyly, and a single palmar crease on the right hand. Additionally, when the thighs were flexed, a curvature was evident subcutaneously at the sacral region, which turned into a caudal appendage upon dissection (9).
When we retrospectively reviewed the anomaly scan images of our patient, we noticed an echogenic double line in the fetal lumbosacral region that strongly resembled the tail observed postnatally. However, the image showed it to be so short that it was unclear whether it had been longer during the antenatal period and was simply misdiagnosed, or if it had elongated later in the antenatal period.
In our case, due to a history of NTD in a previous pregnancy and consanguineous marriage, genetic consultation was conducted, followed by whole exome sequencing. The fetal karyotype was normal, but some phenotype-relevant and irrelevant pathogenic variants were detected. To the best of our knowledge, there has not been a similar report in the English literature. However, Vijayaraghavan et al. introduced a female infant with a lumbosacral tail-like structure and limb defects whose cytogenetic profile was normal (10).
In the current case, all phenotype-relevant variants were of uncertain significance, and one pathogenic genetic variant associated with cutaneous albinism was found. The baby was heterozygous for the mutation, and because of the autosomal recessive inheritance pattern, no apparent phenotypic features were evident until two years of age. Since there are no similar reports regarding WES findings or associated albinism in previously reported cases, the clinical implications remain unclear.
On the other hand, it has been documented that HOX genes determine which embryonic regions develop into cervical, thoracic, lumbar, and sacrocaudal parts. Studies in mouse models have shown that posterior HOX genes play a key role in determining tail length in mammals. For example, in mice, Hoxb13 is related to tail elongation (11, 12). Our case had a mutation in the HOXA13 gene (NM_000522: exon1:c: G196T). The detailed developmental process of regression of the human embryonic tail has not been clearly determined. Some authors have studied human embryonic specimens to evaluate the transition of somites, clarifying that the number of caudal somites decreases significantly immediately after axial elongation. However, the cellular mechanisms and genes involved have yet to be clarified (12).
3.1. Conclusions
In summary, although some genetic variants of uncertain significance and a pathogenic variant associated with albinism were found in our case, the absence of manifestations other than the tail and the lack of literature in this regard make the clinical implications of genetic testing in human tails unclear and warrant further investigation. Moreover, the reported variations were discovered only through the exome method. We suggest validating the identified variations, and other molecular techniques, such as PCR-Sanger sequencing, should also be applied.