
Introduction
Drugs that interact with benzodiazepine receptors have useful effects on treatment of sleep disorders, anxiety, seizure cases and skeletal muscle cramps (1-3). The structure-activity relationship of benzodiazepine receptor ligands has been the subject of many researches. It has been clear that at least two common features are necessary for binding benzodiazepine receptor: an aromatic ring and a coplanar proton-accepting group in a suitable distance. Moreover, the presence of a second out-of-plane aromatic ring could potentiate binding to the receptor especially for agonists (4-8). In our previous studies, we reported some new simple non-rigid derivatives of triazole and oxadiazole rings (9-18), with above-mentioned requirements for binding to the benzodiazepine receptor. Since all of our previously effective reported phenoxyphenyl oxadiazoles had halogen substitute on at list one of the phenyl rings, we synthesized a few phenoxyphenyl-1,3,4-oxadiazole derivatives, structure 1 (Figure 1), without any substitute on the phenyl rings to elucidate the role of electron withdrawing halogen in the benzodiazepine effects. Conformational analysis followed by superimposition of energy minima conformers on known benzodiazepine agonist, estazolam, was performed to clarify whether the synthesized compounds could mimic agonist structure of benzodiazepine ligands. Anticonvulsant activity of the synthesized compounds was determined as an in-vivo model for evaluating benzodiazepine effect.
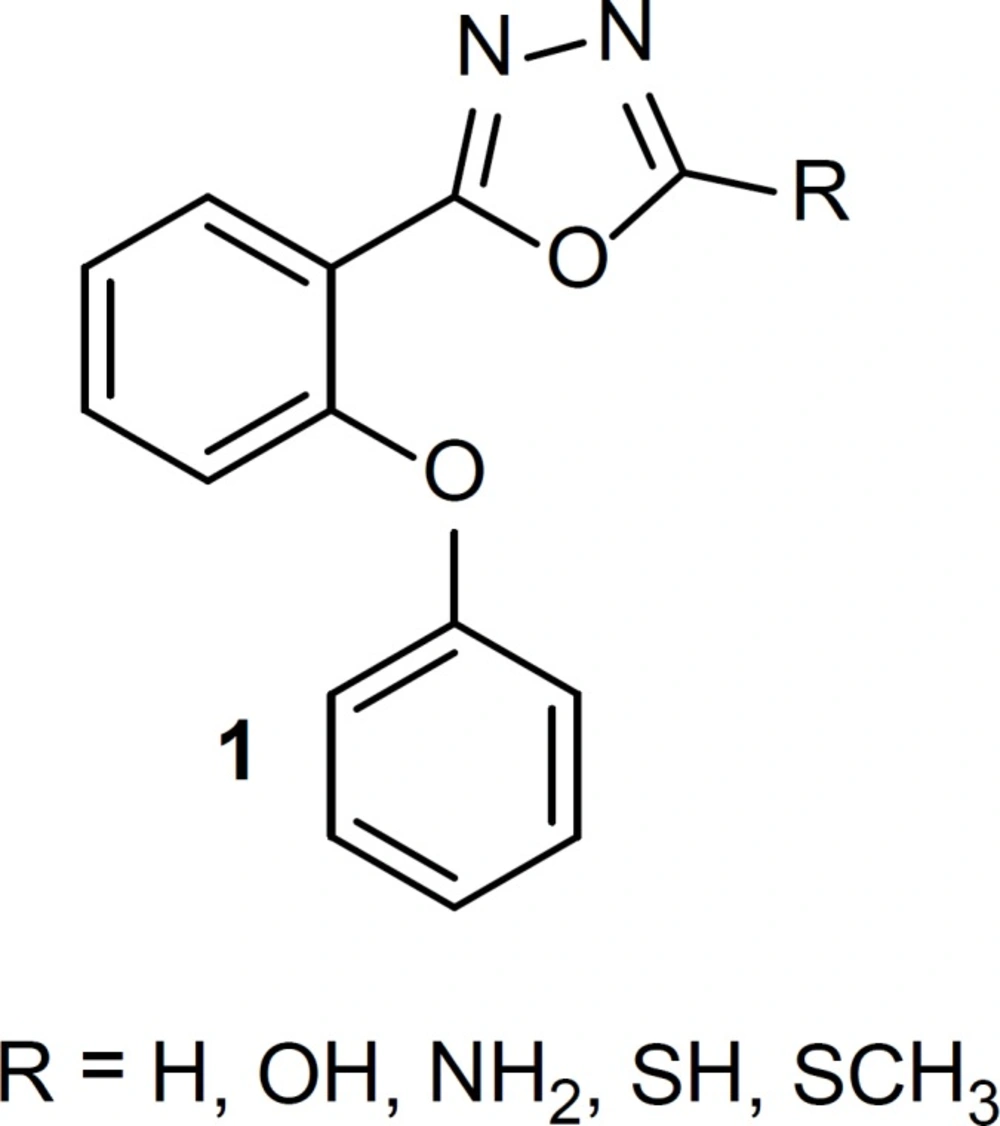
Figure 1
Structure of 1,3,4-oxadiazole derivatives, designed as benzodiazepine receptor ligands
Experimental
Chemistry
Melting points were determined with a Reichert-Jung hot stage microscope and are uncorrected. 1H-NMR spectra were obtained with a Bruker 80 MHz spectrometer.
Chemical shifts are reported in ppm (d) values relative TMS as internal reference. Mass spectra were recorded by a Finnigan Mat TSQ-70 spectrometer. Thin-layer chromatography was carried out using Merck HF254 Silicagel. 2-Phenoxybenzoic acid was purchased from the Fluka Chemie AG. The other chemicals for synthesis were supplied from Merck (Germany).
2-(2-Phenoxy)phenyl-1,3,4-oxadiazole-4(H)-5-tion (6)
To a stirring solution of 4 (3.5 g , 15.4 mmol) and potassium hydroxide (1 g , 18 mmol) in ethanol 96% (85 mL) cooled in an ice bath, carbon disulfide (2.8 g, 36.8 mmole) was added drop wise. The mixture was refluxed for 7 h. The solvent was evaporated and to the residue, water was added and acidified with dilute hydrochloric acid. The precipitate was filtered and crystallized from ethanol 96% to give 3.5 g (85%) of compound 6, mp 199-201°C. IR (KBr): = 3190 cm-1 (NH), 1350 (C=S); 80 MHz 1H-NMR (CDCl3): δ (ppm) 6.80-7.50 (m, 8H, aromatic), 8.10 (dd, 1H, J5,6 = 7.1, J4,6 = 2.1 Hz, phenyl H6); MS : m/z (%) 271 (M+ + 1, 85), 210 (55), 197 (15), 181(100), 152 (20), 77 (5).
2-Methylthio-5-(2-phonxy)phenyl-1,3,4-oxadiazole (7)
To a solution of compound 6 (1 g, 3.7 mmol) in ethanol 96% (2 mL), aqueous solution of NaOH 10% (2 mL) and methyl iodide (0. 53 g , 3.73 mmol) was added and ultrasonicated for 6 min. Water was added and the solution was extracted with chloroform. The organic layer was washed with water and saturated solution of NaCl and dried with sodium sulfate and evaporated to give 1 g (95%) of compound 7 as an oil. 80 MHz 1H NMR (CHCl3-d1) : δ 2.58 (s, 3H, SCH3), 6.80-7.60 (m, 8H, aromatic), 8.05(dd, 1H, J5,6 = 7.2, J4,6 = 2.2 Hz, phenyl H6). MS: m/z (%) 2.85 (M+ + 1, 100), 210 (5), 182 (20), 168 (10), 92 (5), 77 (10).
5-(2-Phenoxy) phenyl-1, 3,4-oxadiazole-3(H)-2-one (8)
To a stirring solution of compound 4 (3 g, 13.2 mmol) and triethylamine (3 mL) in dry tetrahydrofuran (150 mL), cooled in an ice bath, 1,1′-carbonyldiimidazole (4.2 g, 26.3 mmol) was added. The stirring was continued for 5 h. Triethylamine (1.5 mL) and 1,1′-carbonyldiimidazole (1.5 g, 9.4 mmol) were added and stirring was continued at room temperature overnight. The solution was evaporated and the residue was dissolved in ether and washed with hydrochloric acid 20%. A precipitate was appeared witch was filtered and crystallized from ethanol 96% to give 2.1 g (63%) of compound 8, mp 180-183°C. IR (KBr): ν = 3100 cm-1 (NH), 1790 (C=O); 80 MHz 1H-NMR (CDCl3): δ (ppm) 6.85-7.55 (m, 8H, aromatic), 8.12(dd, 1H, J5,6 = 7.0, J4,6 = 2.2 Hz, phenyl H6). MS: m/z (%) 254 (M+, 100), 197 (35), 152 (15), 120 (40).
2-Amino-5-(2-phenoxy) phenyl-1,3,4-oxadiazole (9)
A solution of compound 4 (6 g, 26.3 mmol) and cyanogen bromide (3.1 g, 29.3 mmol) in methanol (100 mL) was refluxed for 2 h. The solvent was evaporated and the residue was crystallized from ethanol 96% to give 5.3 g (80%) of compound 9, mp 212-214°C. IR (KBr): ν = 3280 and 3200 cm-1 (NH2), 3120 (NH dimer), 1660 (C=N); 80 MHz 1H-NMR (CDCl3): δ (ppm) 5.6 (broad s, 2H, NH2), 6.80-7.50 (m, 8H, aromatic) 8.00 (dd, 1H, J5,6 = 7.2, J4,6 = 2.1 Hz, phenyl H6); MS : m/z (%) 253 (M+, 100), 210 (10), 181 (35), 152 (10), 115 (5), 77 (5).
2-(2-Phenoxy) phenyl-1,3,4-oxadiazole (10)
To a stirring solution of compound 5 (2 g, 7.8 mmol) in dry xylene (90 mL), P2O5 (4 g) was added and refluxed for 2 h. The solution was decanted and evaporated. The residue was purified by thin layer chromatography (chloroform-methanol, 98:2) to give 1.2 g (65%) of compound 10, mp 60-62°C (ether). 80 MHz 1H-NMR (CDCl3): δ (ppm) 6.80-7.60 (m, 8H, aromatic), 8.14 (dd, 1H, J5,6 = 8.0, J4,6 = 1.6 Hz, phenyl H6), 8.45 (s, 1H, oxadiazole); MS: m/z (%) 238 (M+, 100), 221 (65), 180 (55), 152 (40), 138 (5), 91 (25), 77 (35), 51 (50).
Conformational analysis
Conformational analysis of the designed compounds was performed by MM+ force field followed by AM1 method implemented in HyperChem 7 software (Hypercube Inc.). Global minimum forms of the compounds were superimposed on estazolam which was considered as a reference.
Pharmacology
Male NMRI mice (Pasteur Institute Iran) weighting 20-25 g (n = 10) were used in the experiments. The animals were kept in groups of ten in cages under constant temperature ( 24 ± 1°C ) and 12 h light/dark schedule. They had free access to standard mouse diet and tab water except during the experiment. On the day of the experiment, animals were transferred to individual cages randomly and allowed to acclimatize for 30 min before injection of drug or vehicle. Anticonvulsant activity of compounds was measured by PTZ-induced lethal convulsion test (21).
Test compounds and vehicles were administered to groups of ten mice 1 h before the injection of PTZ (100 mg/Kg, ip) and the number of dead mice were counted 30 min after injection of PTZ.
Test compounds and flumazenil (Hoffmann La Rosch) were suspended in a solution of carboxymethylcellulose (CMC) (1%) and tween 80 (0.5%) in distilled water. Diazepam ampoules (Darou Pakhsh Co.) were diluted with distilled water. Test compounds, diazepam and flumazenil were injected 1 h, 30 min, 15 min before injection of PTZ respectively. Control animals were given the corresponding vehicles. This study was conducted in accordance with protocols approved by the Institutional animal care and use committee and all experiments were performed based on the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and all efforts were made to minimize the number of animals used in the study.
ED50 values and their 95% confidence limits were determined by computer programmed probit-regression. Fisher’s exact probability test was used to compare flumazenil treated and non-treated groups. All the data were presented as Mean (95% confidence limits) and p < 0.05 considered statistically significant.
Results
Chemistry
1, 3, 4-Oxadiazole derivatives 6-10 were synthesized according to Figure 2.
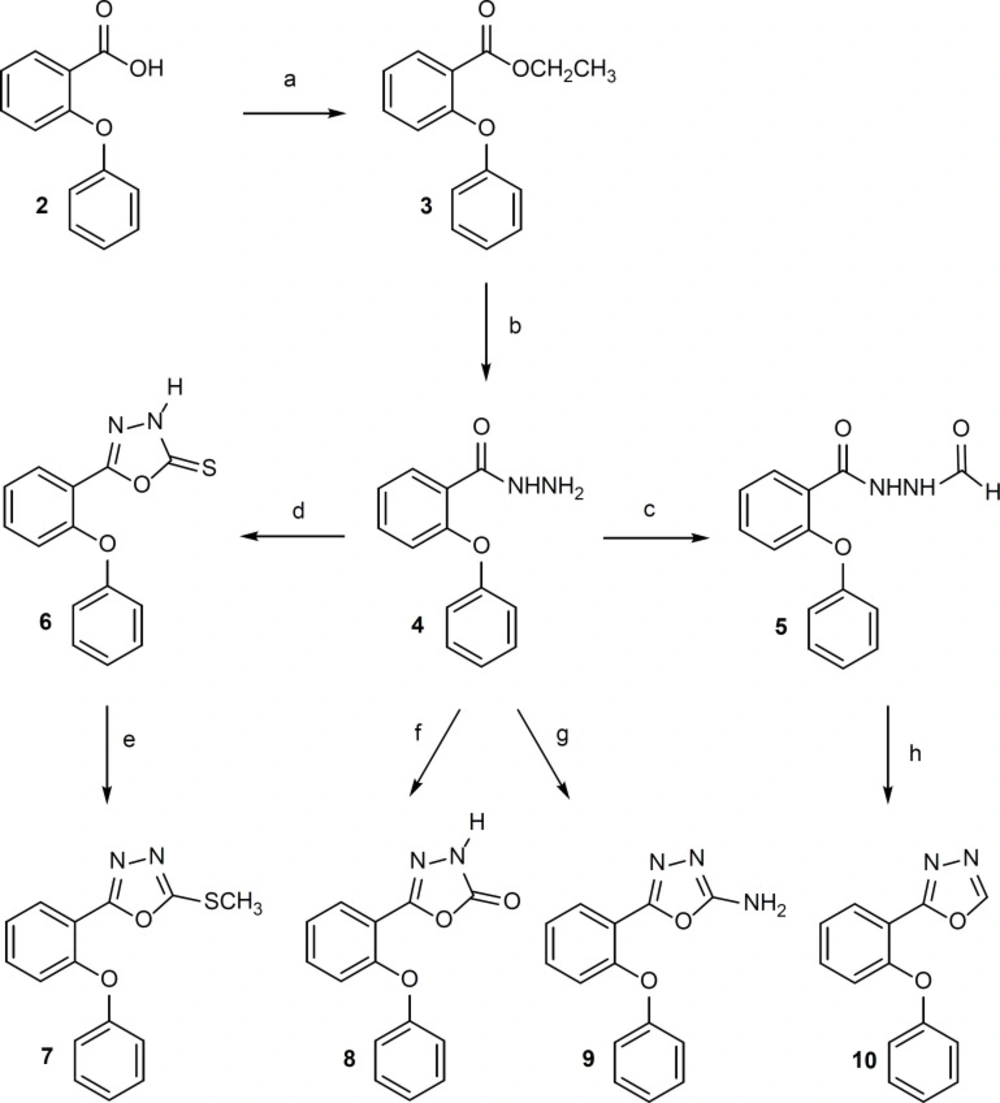
Figure 2
Reagents: (a) ethanol, concentrated H2 ; (d) CS2, ethanol 96%, KOH, reflux, 7 h; (e) CH3I, NaOH 10%, ethanol 96%, ultrasonication, room temperature, 6 min; (f) 1,1′-carbonyldiimidazole, triethylamine, dry THF, 0°C, 5 h, room temperature, 12 h; (g) BrCN, methanol, reflux, 2 h; (h) P2O5 , xylene, reflux, 2 h.
Compound 3 was obtained in quantitative yield by esterification of 2-phenoxybenzoic acid with ethanol. Compound 4 was prepared in satisfactory yield from the reaction of 3 with hydrazine hydrate in ethanol at room temperature. Compound 5 was synthesized by refluxing of 4 in formic acid. 1,3,4-Oxadiazole-4(H)-5-tione ring (Compound 6) was synthesized through refluxing of 4 with carbon disulfide in alcoholic caustic potash. Methylation of 6 with methyl iodide gave 7 in high yield. Compounds 8 and 9 were prepared in good yield by the reaction of 4 with 1,1′-carbonyldiimidazole and cyanogen bromide respectively in proper conditions. Dehydration of 5 by phosphorus pentoxide in boiling toluene gave 10 (10, 18-20).
Conformational analysis
Since compounds 1 (Figure 1) are non-rigid structures, conformational optimization with MM+ force field followed by AM1 calculation was performed to confirm whether these compound could mimic proper conformation for binding to the benzodiazepine receptors. A known benzodiazepine agonist, estazolam, was considered as a reference compound and so the procedure was also performed on that.
Superimposition of the energy minima conformers of the designed compounds on estazolam was performed to examine the matching of the benzodiazepine pharmacophores, aromatic rings and proton accepting groups (Figure 3).
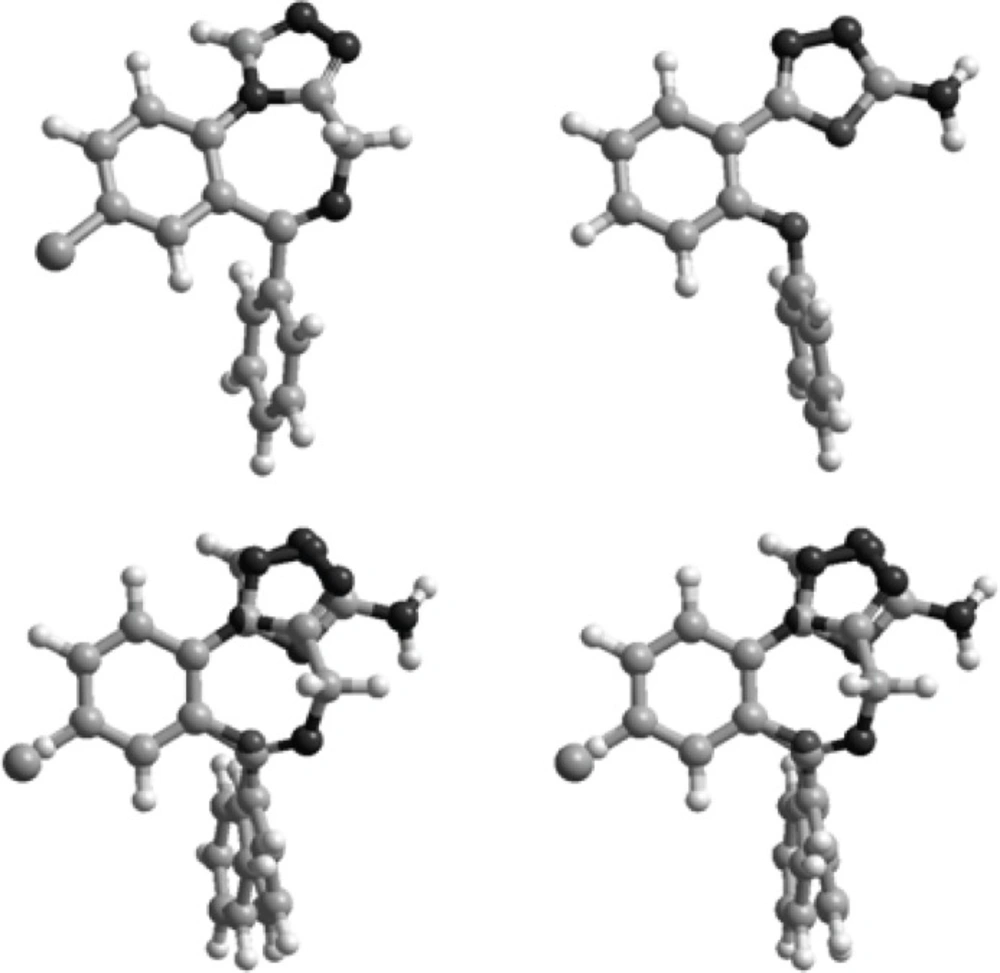
Figure 3
Stereoview of the superimposition of the energy minima conformers of estazolam (left) and compound 9 (right).
Pharmacology
Anticonvulsant effect of compounds was determined as a model for benzodiazepine activity through evaluation of the ability of the synthesized compounds to protect mice against a lethal dose of pentylenetetrazole (PTZ) (Table 1).
Table 1Anticonvulsant activity of compounds 6-10 in PTZ-induced lethal convulsion test
To confirm whether the anticonvulsant activity of the compounds mediates through benzodiazepine receptors, the effect of flumazenil, a benzodiazepine antagonist, in reducing the anticonvulsant activity of the compounds was determined.
Discussion
In our previous studies, we reported phenoxyphenyl oxadiazoles with halogen substitute on at list one of the phenyl rings. In the present work, we synthesized a few phenoxyphenyl-1,3,4-oxadiazole derivatives 6-10 without any substitute on the phenyl rings to elucidate the role of electron withdrawing halogen in the benzodiazepine effects. Table 1 shows the anticonvulsant activity of compounds 6-10. Diazepam was considered as a standard benzodiazepine agonist. The results show that compound 9 with an amino substituent on 2 position of 1,3,4-oxadiazole ring has a respectable anticonvulsant activity and replacement of this group with H, OH, SH or SCH3 abolish or decrease the anticonvulsant effect. The fact that the activity of compound 9 is significantly reduced by flumazenil, a known benzodiazepine antagonist, confirms that this effect is mediated through benzodiazepine receptors.
Figure 2 illustrates the superimposition of energy minima conformers of compound 9 on estazolam, a known benzodiazepine agonist. It is clear that the aromatic rings and proton accepting groups, N-3 of the oxadiazole ring of compound 9 and N-2 of the triazole ring of estazolam, are well matched. These groups could be considered as the main proposed benzodiazepine pharmacophores. Therefore, compound 9 could mimic the benzodiazepine structure at the receptor sites. These results are entirely consistent with our previous findings on the other 1,3,4-oxadiazole and 1,2,4-triazole derivatives (9-18). Although compound 9 is weaker than diazepam, it should be mentioned that it has a good margin of safety and LD50 of this compound is bigger than 15 times of its ED50 (Table 1). The results are in agreement with SAR of benzodiazepine receptor ligands because in the present work we eliminated the electron withdrawing substituent in position 2 of phenoxy ring and position 4 of phenyl ring of our previously designed molecules and the anticonvulsant activity reduced. This finding confirms that the designed structures bind to the receptors as the same manner of the classic benzodiazepines.
