










1. Introduction
Inherited myopathies are inherited disorders known by progressive muscle weakness and are very similar and yet heterogeneous in their clinical, pathological, and genetic classifications. However, the maximum joint separation is formed based on the onset age and the concerned (e.g., facial, proximal, and distal) muscles (1, 2).
Although many inherited dystrophies are associated with the dysfunction of dystrophin and its related proteins, this is not a specific mechanism for inherited muscular dystrophy. A variety of cellular pathways are associated with disorders related to inherited muscle diseases (3). Examples are summarized in Table 1.
| Gene product | Location | Phenotypes |
|---|---|---|
| Emerin | Nuclear envelope | X-EDMD |
| Lamin A/C | Nuclear envelope | AD-EDMD LGMD 1B |
| Dysferlin | Membrane | Distal myopathy LGMD 2B |
| Caveolin 3 | Caveolae | LGMD 1C |
| Calpain 3 | Cytoplasm | LGMD 2A |
| Integrin α7 | Membrane | Congenital myopathy |
| Collagen VI | ECM | Bethlem myopathy |
| DMP | Neuromuscular junction | Myotonic dystrophy |
| ZNF9 | Nuclear | PROMO |
| PABP2 | Nuclear | OPMD |
| Telethonin | Sarcomeric | LGMD 2G |
| Myotilin | Sarcomeric | LGMD 1A |
Abbreviations: EDMD, Emery-Dreifuss muscular dystrophy; LGMD, limb-girdle muscular dystrophy; DMPK, myotonic dystrophy protein kinase; PROMM, proximal myotonic myopathy; OPMD, oculopharyngeal muscular dystrophy; and PABP2, polyA-binding protein 2.
Breaking the link between the cytoskeleton and the extracellular matrix is understood to be a concern within the pathological process of a variety of transmissible muscle diseases, rare collagen VI–related disorders, autosomal recessive myosclerosis, Bethlem myopathy (BM), Ulrich congenital muscular dystrophy (UCMD), autosomal dominant limb-girdle muscular and dystrophy phenotype (4-7).
Inherited myopathies present in childhood with variable symptoms such as hypotonia, feeding and metabolic process difficulties, and delayed motor skill development. Some patients with inherited less severe disease may present scoliosis, contraction, and respiratory failure later in life (1). Typically, clinical findings include general or proximal weakness, hypotension, muscle weakness, and secondary deformity due to muscle weakness. Usually, the diagnosis of inherited myopathies is made upon a detailed history and examination, followed by biochemical and neuroscience assessment and completed by muscle biopsy and genetic testing (5).
Although the involved muscles are sometimes concerned, the involvement of cardiac muscles is rare. Electromyography is ordinary and can show low-amplitude, polyphasic (myopathic) potentials. Changes specific to the myopathy are visible through muscle biopsy without fibrotic or necrotic changes (5, 8).
Bethlem and Wijngaarden were the first to discover BM in 1976 (5). Normal flexion contractions, slowly progressive weakness of the proximal muscles, flexors of the long fingers, wrists, elbows, pectoralis major, and ankle dystrophy are characteristic of this myopathy. Patients with BM sometimes become symptomatic with hypotonia at birth. However, these patients will usually show symptoms in the first or second decade of life. Patients with BM continue walking until adulthood, but two-thirds of them need walking aid at the age of about sixty. The decline in respiratory function is variable and does not occur until adulthood in these patients (9). Noticeably, UCMD is a more severe form of the disease, with congenital weakness and tone, proximal joint contractions, and excessive loosening of the distal joints. Unlike BM, where respiratory involvement is relatively rare, patients with UCMD usually need respiratory support (10).
In UCMD, the onset is usually birth or during the preceding year of life with a delay in acquiring motor skills. Patients with UCMD never begin walking or, if they do, they will lose their ability to walk around the age of ten. A decline in pulmonary function occurs early, and thus beginning mechanical ventilation at night from the first or second decade of life is very important and helpful (11). The prevalence of BM and UCMD is reported to be 0.77:100,000 and 0.13:100,000, respectively, although this rate may not be precise as some patients could have been underdiagnosed (12). Here, we reported clinical and molecular genetic findings in four unrelated families.
2. Cases Presentation
2.1. Patient 1
The proband (P1) belonged to a 6-year-old symptomatic boy with myopathy. The patient’s parents were relatives. He had had a history of proximal muscle weakness since childhood. Creatine kinase levels mildly increased (about 540 U/L) in the patient, and his electromyography showed a myopathic pattern. There was history of affected individuals in their family with the same symptoms such as difficulty in walking, speech, and limb deformity.
2.2. Patient 2
The proband (P2) represented a 10-year-old boy. The patient’s disease symptoms started at the age of five, including severe muscle atrophy and difficulty in walking and standing up. The patient’s electromyography showed a myopathic pattern. Creatine kinase levels moderately increased (about 4000 U/L) in the patient constantly. In his examination, the muscular atrophy of the distal and proximal limbs was observed. The patient’s father was also affected and had his son’s exact symptoms.
2.3. Patient 3
The proband (P3) was a 17-year-old boy with high CK (2592 UI/L) at the age of 14 who showed mild axial muscle weakness. He had trouble breathing, difficulty in standing up, fatigue, speech problems, and poor vision but had no heart problems. Creatine kinase levels moderately increased (about 2952 U/L) in the patient, and his electromyography showed irritable myopathy. The proband’s 8-year-old sister presented with a similar clinical history and a milder phenotype: she could barely stand up and had difficulty breathing in her sleep. Also, one of her lower limbs was shorter than the other.
2.4. Patient 4
The proband (P4) was a 28-year-old male with creatine kinase levels of about 4708 U/L and normal echocardiography. The patient’s muscle biopsy results showed that the striated muscles were involved in necrosis and atrophy in a non-fascicular manner. The proband walked with difficulty and the help of others. He also had progressive paralysis, and his sister and cousin had similar but milder symptoms.
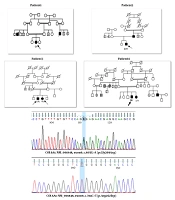
The family pedigrees and main clinical manifestation of all the patients in the present study are shown in Figure 1 and Table 2, respectively.

The pedigrees of reported families. Black shapes show affected individuals. The chromatogram of validated variants through Sanger sequencing.
| Patient | Age (y) | Onset (y) | sex | Max CK (U/L) | Pattern of Contractures | Severity |
|---|---|---|---|---|---|---|
| P1 | 6 | < 2 | Female | 540 | Proximal weakness | Mild |
| P2 | 10 | > 4 | Male | 4000 | Moderate proximal. Distal laxity | Moderate |
| P3 | 17 | > 12 | Male | 2952 | Moderate weakness | Moderate |
| P4 | 28 | > 14 | Male | 4708 | Proximal contractures, distal hyperextensibility | Severe |
3. Methods
We investigated many patients from different centers and referrals from neurologists to our Genetics Research Center. After genetic counseling, completing the relevant questionnaire, and obtaining consent form for the clinical diagnosis of myopathic diseases, all laboratory procedures, including muscle enzymes, creatine phosphokinase and aldolase, LDH, SGPT, SGOT, MRI, ECG, electromyography, and genetic testing, were briefly used. Five ml peripheral whole blood was collected in an EDTA tube from available family members who took part in genetic tests. The first step in genetic diagnostics is DNA extraction using the Gene All Kit. We performed whole exome sequencing (WES) through the Illumina HiSeq 4000 system (Illumina, Inc., San Diego, CA, USA). FASTQ files were aligned to the human reference sequence (hg19) by the Burrows-Wheeler Aligner, and SAM files were produced.
Further, SAM to BAM conversion, BAM file sorting, and removal of duplicate reads were carried out using Picard (http://picard.sourceforge.net), followed by local realignment and variant calling using the Genome Analysis Tool Kit to generate VCF files. Then, annotation was performed with Annovar32 to identify pathogenic variants in affected individuals. Sanger sequencing was done using specific designed primers to confirm the candidate variants in patient and their parents.
3.1. Molecular Findings
The analysis results of COL6A genes in all patients included in the present study are summarized in Table 3. We detected a heterozygous variant in the exon9 of the COL6A1 gene c.805G>A (p.Gly269Arg) in Patient 1. This variant has been described as the primary etiology for Ulrich's congenital muscular dystrophy (autosomal recessive and autosomal dominant). It is classified as a likely pathogenic variant (class 2) according to the recommendation of the American College of Medical Genetics and Genomics guideline. The parents and other family members should consider genetic counseling and confirmation of the variant by Sanger sequencing. Patient-4 revealed a homozygous mutation in the exon8 of the COL6A1 gene c.784C>T (p.Arg262Trp) and showed the limb-girdle muscular dystrophy (LGMD) symptoms. One heterozygous mutation in the exon 15 of the COL6A3 gene c.5581G>A (p.Glu1861Lys) was reported in Patient-2, although its frequency is very low in the average population.. BM 1 and Ulrich congenital muscular dystrophy 1 are inherited in an autosomal recessive/dominant manner. In Patient-3, a homozygous mutation was detected in the exon28 of the COL6A2 gene (c.2917G>A, p.Val973Met). Also, a homozygous variant was detected in the exon8 of the COL6A1 gene (c.784C>T, p.Arg262Trp) in Patient-4. This gene has been reported in association with autosomal recessive Ulrich's congenital muscular dystrophy1.
| Patient | Gene | Ref | Nucleic Acid Alternation | Amino Acid Alternation | Exon | Heredity | Zygosity | Insilco Prediction |
|---|---|---|---|---|---|---|---|---|
| P1 | COL6A1 | NM_001848 | c.805G>A | p. Gly269Arg | 9 | AR, AD | Het | likely pathogenic |
| P2 | COL6A3 | NM_057167 | c.5581G>A | p. Glu1861Lys | 15 | AR, AD | Het | likely pathogenic |
| P3 | COL6A2 | NM_001849 | c.2917G>A | p. Val973Met | 28 | AR, AD | Hom | VUS |
| P4 | COL6A1 | NM_001848 | c.784C>T | p. Arg262Trp | 8 | AR, AD | Hom | VUS |
Abbreviations: Het, heterozygous; Hom, homozygous; AR, autosomal recessive; AD, autosomal dominant; VUS, variant of unknown significance.
4. Discussion
Col-VI consists of three chains (alpha1, alpha2, and alpha3) encoded by totally different genes (COL6A1, COL6A2, and COL6A3). These three chains act within the cell and make a triple-helical compound assembled in dimers and tetramers stabled by disulfide bonds. Each chain contains a relatively short triple helical domain with repetition Gly-X-Y subunits, flanked by sizeable globular N- and C-terminal domains (13, 14). The α1 (VI) and α2 (VI) chains are estimated to be around 1,000 amino acids long and have a molecular weight of 140-150 KDa. However, the α3 (VI) chain length is more significant as it contains numerous particles in the range of 2,500 to 3,100 amino acids and has a molecular weight of 250-300kDa (5, 15).
Collagen VI (Col-VI) is a protein extensively used in the extracellular matrix (ECM) of most connective tissues that help maintain tissue integrity and function (16). This protein is also considered a critical member of energy metabolism in adipose tissue, and low serum levels of this protein are reported related to fat distribution (15, 17). Col-VI is the main protein in the structure of multiple organs and tissues such as lungs, brain, myocardium, peripheral nerves, blood vessels, intervertebral disc, cornea, and skin (17).
Collagen VI-related disorders were first clinically recognized, and their pathophysiology was identified long after. These disorders have a range of different phenotypes, including BM, UCMD, limb-girdle muscular dystrophy, and myosclerosis myopathy. Some of these phenotypes may have overlapping clinical manifestations, but they have a different course of management, and this is a strong reason for specifying their clinical definitions (6, 7, 18).
According to muscle biopsy results, it is known that dystrophic features can be subcategorized to into degeneration, regeneration, and replacement of muscle tissue more often with fat tissue. Due to the genetic heterogeneity of the mentioned myopathies, the detection rate is relatively low. There have been some solutions, such as deletion/duplication analysis, but studies have shown no improvement in the detection rate. The coding region sequencing of the three genes (COL6A1, COL6A2, and COL6A3) has been accentuated by many reliable studies to confirm the detection of two mutant alleles (9, 19).
In conclusion, we found four different genetic variants in the COL6A1, COL6A2, and COL6A3 genes through a clinical genetic study. As a practical approach, NGS provides information for genetic counseling of related families. We hope that these findings help understand collagen VI-related myopathies.
