





1. Background
Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are X-linked recessive neuromuscular disorders characterized by progressive muscle fibers degeneration and weakness of skeletal muscles (1). With an incidence of 15.9 - 19.5/100000, DMD/BMD is the most common inherited muscular dystrophies in childhood (2). They are caused by mutations in the dystrophin gene, which leads to the absence or deficiency of the dystrophin protein (3, 4). Dystrophin is one of the largest human genes, located on the short arm of the X chromosome near the region Xp21 (5). It consists of 79 exons that encode a 14 kb mRNA, which makes a 527-kDa dystrophin protein (6). The lack of this protein in muscle cells causes early damage and fragility. Moreover, in the progressive form, loss of dystrophin function leads to inefficient fiber regeneration and ultimate replacement of muscle tissues by adipose and connective tissues (1). It has been estimated that approximately one-third of the DMD/BMD cases are due to de novo mutations (7). Studies have indicated that different types of mutations demonstrate a wide range of symptoms with variable phenotypic presentations (3, 4). DMD patients are usually men and rarely women. They begin to show symptoms between the ages of 2 - 5 years (1). Primarily, DMD results in difficulty in walking, jumping, and running, and later it leads to a progressive loss of cardiac and respiratory reserve capacity.
Eventually, more progressive weakness and scoliosis may cause respiratory failure and insufficiency, wheelchair dependency, and heart problems, which ultimately result in death (8). Duchenne muscular dystrophy carriers are females who may present with classic dystrophinopathy or may be asymptomatic throughout their lives. They have one abnormal X chromosome for dystrophin and a 50% risk of giving birth to male offspring with the disease (9). Becker muscular dystrophy is similar to DMD; however, the onset is usually in the teens, and its clinical course and progression are milder and slower (1). Broad types of mutations extending from large deletions and duplications to small point mutations (frameshift, nonsense, and missense types) have been linked to DMD/BMD (10). Intragenic deletions or duplications along the dystrophin gene have been observed in nearly 70% of the patients (especially exons 44 to 53) (11, 12). In the remaining 30% of DMD/BMD cases, small point mutations in the absence of large deletion or duplication are detected (11).
Since exonic deletions or duplications are the predominant types of pathogenic variants in the dystrophin gene, an initial quantitative method, which detects the majority of these copy number variations, should be the first diagnostic test offered (13). Multiplex ligation-dependent probe amplification (MLPA) is a simple and robust method for the detection of large intragenic deletions and duplications in affected patients and carrier females (14). However, MLPA only determines the extent of deletions or duplications to the exon level. It is unable to detect unknown point mutations and small deletions (10, 15). Therefore, if the MLPA test results were negative, first-generation sequencing, known as the Sanger method and NGS, are powerful methods, which should be considered (16-18).
2. Objectives
In the current study, we aimed to assess mutations in subjects with clinical suspicion of DMD/BMD using MLPA as the first-line detection method. Then, NGS and Sanger sequencing techniques were applied in MLPA negative cases.
3. Methods
3.1. Subjects
Thirteen clinically suspected DMD/BMD cases who were referred for genetic testing to Genome Genetic Laboratory, Tehran, Iran, from August 2019 to August 2020 were included in this study. The ethics committee of Tehran Medical Sciences, Islamic Azad University approved the study protocol (IR.IAU.PS.REC.1399.070), and all participants or their legal guardians signed the written informed consent.
Our inclusion criteria were subjects who had clinical manifestations of progressive muscle weakness and were clinically suspected of DMD/BMD. For all families, pedigree analysis and genetic counseling were performed in the first and the following visits by a single trained physician.
The peripheral blood samples were collected from all subjects and evaluated by the MLPA, NGS, and Sanger techniques in the aforementioned lab.
In the first step, MLPA was performed to detect deletions and duplications in all patients. Patients with negative MLPA results were examined for small mutations using NGS and Sanger sequencing.
3.2. Definition of Techniques
3.2.1. DNA Extraction
DNA was extracted from blood samples by applying the salting-out method (19). All DNAs were quantified by Nanodrop (Thermo Scientific™ NanoDrop™ One) and analyzed for the degradation on the agarose gel. Degraded DNA was not employed for MLPA analysis.
3.2.2. Multiplex Ligation-Dependent Probe Amplification Assay
Multiplex ligation-dependent probe amplification was performed using the P034/P035 DMD Kit (MRC-Holland, Amsterdam, Netherlands) according to the manufacturer’s instructions. The resulting fragments were separated using ABI PRISM 3100 (ThermoFisher Scientific-USA) and analyzed by GeneMarker software version 1.95. Peak heights were normalized to control healthy individuals, and a deletion or duplication was expected when the normalized peak ratio value was 0 or 2 for male patients. In female cases, values of 1, 3, and 4 indicated heterozygote deletion, heterozygote duplication, and heterozygote triplication, respectively (15).
3.2.3. Next-Generation Sequencing
Next-generation sequencing covering the dystrophin gene was operated on about 25 ng of dna. Two primer pools were arranged by Ion Ampliseq Library Kit 2.0-96 LV (cat.448044). For DNA libraries with two primer pools target, amplification reactions were combined. Ligation of adapters to the amplicons and their purification were done by Ion Xpress TM P1Adaptor, and then the library was purified by AMPure XP reagent followed by equalization of the library with Ion library Equalize KIT (cat: 4482298).
Amplifion of the library, washing the Equalizer TM Beads, adding Equalizer™ Capture to the amplified library, combining captured libraries, adding Equalizer™ Beads and wash, and eluting the Equalized library were performed by Ion Torrent instrument (ThermoFisher Scientific -USA) according to the manufacturer’s protocol. To validate the mutations identified with NGS, Sanger sequencing was done on 3500 Genetic Analyzer (ABI, Carlsbad, CA, USA).
3.3. Statistical Analysis
Descriptive statistics were used to assess the baseline characteristics (means (SD) and numbers (proportions). Statistical analysis was performed using SPSS software (version 16, SPSS Inc., Chicago, IL, USA).
4. Results
4.1. Demographic Data
Thirteen clinically suspected DMD/BMD cases (nine males and four females) were recruited for the current study. The age range of the subjects was 2 - 40 years (mean ± SD): 16 ± 7.921 years old). Table 1 demonstrates the demographic and clinical data of all studied subjects. Positive family history of DMD/BMD was presented in 23.077% of the cases who were female. Consanguineous marriage was observed in 38.46% of the patients.
| Sample ID | Age | Sex | Clinical Presentation | Family History of Duchenne Muscular Dystrophy/Becker Muscular Dystrophy | Relative Marriage | Other Familial Genetic Disorder |
|---|---|---|---|---|---|---|
| M98-275 | 7 | Male | Problem in walking since five years ago; muscle deterioration and muscle atrophy; high liver enzymes; elevated CK concentration,abnormal EMG. | Negative | Positive | Negative |
| M99-635 | 7 | Male | Inability to go upstairs and walk since one year ago. | Negative | Negative | Negative |
| M98-9 | 27 | Male | Progressive proximal muscle weakness; calf hypertrophy since 12 years ago. | Negative | Negative | Negative |
| M98-2188 | 10 | Male | Muscle weakness; severe muscle atrophy | Negative | Negative | Negative |
| M99-683 | 10 | Male | Inability to go upstairs and to arise since three years ago; muscle weakness of proximal thighs. | Negative | Negative | Negative |
| M98-2375 | 23 | Female | Muscle weakness. | Negative | Negative | Negative |
| M98-400 | 30 | Female | No symptoms and signs. | Positive | Negative | Negative |
| M98-2369 | 24 | Female | No symptoms and signs. | Positive | Positive | Negative |
| M98-655 | 12 | Male | Waddling gait and Gower’s sign; problem of walking and sitting; elevated liver enzymes. | Negative | Positive | Negative |
| M97-1204 | 2 | Male | Gowers’ sign; Muscle weakness and; elevated CK concentration; elevated liver enzymes; abnormal EMG | Negative | Negative | Negative |
| M98-561; | 11 | Male | Walking problems | Negative | Positive | Negative |
| M99-2519 | 5 | Male | Inability to go upstairs and run Gowers’ sign; abnormal EMG; elevated CK concentration | Negative | Positive | Positive |
| M99-2636 | 49 | Female | Family history of DMD; abnormal EMG | Positive | Negative | Positive |
The Demographic and Clinical Data of the Studied Subjects
4.2. Multiplex Ligation-Dependent Probe Amplification Results
Among 13 subjects who underwent MLPA, DMD/BMD causative mutations were detected in ten cases (Appendix 1 in the supplementary file). Next-generation sequencing followed by conformational Sanger sequencing was performed for three MLPA negative cases.
4.3. Sequencing Results
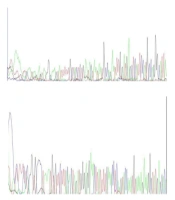
Next-generation sequencing was performed for three MLPA-negative cases. In contrast, analysis of NGS data did not reveal any pathogenic/ likely pathogenic variant in two cases. An 11-year-old boy with progressive walking difficulties and suspicion of DMD had a hemizygous pathogenic variant (c.1836_1840delGAAAA, p.K612*fs) in exon 16 of the dystrophin gene. The result of Sanger sequencing for two negative cases indicated no mutation in the proband (Figure 1).
 results in two negative cases.")
Sequencing analyses to validate next-generation sequencing (NGS) results in two negative cases.
4.4. Frequency of Dystrophin Gene Deletion and Duplication
In the present study, nine exonic deletions were identified by the MLPA method. Deletions were in exons 3 to 10, 21 to 30, 45 to 49, and 11 to 20. The most common multiple exon deletions were 46 to 47 and 4 to 7. Furthermore, heterozygote triplication of exon 71 was found in one female case with muscle weakness and negative family history (Figure 2).

Frequency of dystrophin gene deletion among 36 exons in this study.
In the present study, 76.92% of the patients had intragenic deletions, and 7.7% had intragenic triplication. Analysis of 13 subjects showed that 23.07 % (3 male) of deletions involved a single exon; 30.76% (three males and one female) exhibited deletion of two to three adjacent exons; 15.38% (two males) had contiguous five exons deletion, and 7.7% (one male) more than five exons deletion. Conclusively, 53.84% of the subjects presented multiple-exonic deletions, whereas 23.7% of subjects presented a single-exon deletion. Only one female demonstrated heterozygote triplication (four copies) of exon 71.
5. Discussion
Both DMD and BMD are progressive X-linked recessive genetic disorders caused by mutations in the dystrophin gene. Since dystrophin is one of the largest human genes, it can frequently acquire mutations involving large exonic duplications or deletions (≥ 1 exon) and, to a lesser extent, point, mutations and small deletions (20). However, the prevalence of mutations, such as deletions, duplications, or point mutations is a matter of debate in different studies. Most identified mutations in DMD are large deletions (65%), followed by point mutations (26%), duplications (7%), and others 2% (21, 22). Considering the large size of the gene (> 2.2 Mb) and the wide variety of mutations, the comprehensive molecular diagnosis of DMD/BMD is challenging (23). Also, identification of detailed mutational spectrum is essential for research in specific genetic therapy (20).
For these reasons, a variety of genetic approaches have been applied to study causative mutations in affected patients (11). Among the many quantitative methods available, MLPA is currently the method of choice as an initial diagnostic test in laboratories since it contains a probe for each of the 79 exons and detects single/multiple exonic deletions/duplications simultaneously (24-26). Hence, the reliability of MLPA results is high for copy number variations involving multiple adjacent exons but less in MLPA negative cases that are highly suspected of DMD/BMD. Therefore, investigation of all coding exons for small deletions or point mutations by an independent method (NGS or Sanger sequencing) is recommended by guidelines in MLPA negative subjects (13).
In the present study, the MLPA method indicated causative mutation in ten patients with nine exonic deletions. The deletions were presented mostly in exons 46 to 47 and 4 to 7. These data are consistent with other studied populations (27). Only a heterozygote single exon triplication was detected in a female carrier who presented with muscle weakness and negative family history. Next-generation sequencing was performed as the second diagnostic approach, followed by Sanger sequencing in MLPA negative cases.
In accordance with the previous studies, our findings demonstrated MLPA as an efficient method for the detection of both deletions and duplications of the dystrophin gene. The deletion rate among our Iranian cohort was 76.92%, and the most frequent deletions were the contiguous deletion of exons 46 to 47 and 4 to 7, which is in agreement with previous reports (25, 28-30).
Furthermore, we found two hotspots deletion regions, exon 46 to 47 and exon 4 to 7, with highly distributed mutation, which are in concordance with other studies (31). Also, in comparison with available studies on diagnosis of Duchene disease (1, 10, 11, 28, 32, 33), our result confirmed that deletion mutations occur at a higher frequency compared to duplications.
On the other hand, several investigations on the Middle Eastern populations demonstrated a lower prevalence of deletions compared to our findings (14, 34, 35). These differences between reports can be explained by applied genetic diagnostic techniques, racial and ethnicity variations, geographical distribution, and inclusion criteria. Besides, the pattern of dystrophin gene deletions in our subjects was in accordance with previous studies in Iran and other populations showing multiple-exonic deletions (14, 36-39).
In their study, Zamani et al. reported that in Iranian DMD patients, the most frequent genetic mutations were deletions (nearly 80%), mostly positioned within two hotspots of the dystrophin gene and cluster around specific exons (exons 1 to 20 and 44 to 55) (10). However, the duplication mutations were indicated to be less common in the similar exons (10). Besides, while deletions were predominant in exons 44 to 55, duplications were mostly located in exons 1 to 20 (10). Point mutations (missense, nonsense, splicing, pseudoexon, and frameshifts) were detected in nearly 14 % of the subjects by the Sanger method (40).
In accordance with the current study and Zamani et al.’s report (10), the frequency of dystrophin gene deletions was reported to be 70 - 80% in the European population (6, 10, 30, 41). In contrast, a study on Spanish and Chinese DMD cases demonstrated that the rate of large deletions was 46.1% and 50% in the dystrophin gene, respectively (21, 42).
Currently, large numbers of studies are being conducted to develop optimal treatments and genetic-based therapeutic strategies for DMD/BMD patients, such as gene substitution, gene correction, or reform gene productions. These goals would be achieved only by recognition of precise types of mutations. Thus, our findings would support potential therapeutic targets for DMD/BMD treatment.
5.1. Conclusions
In this study, the number of deletions was more than other mutations in Duchenne/Becker muscular dystrophy. Most deletions were demonstrated in two hotspot regions from exons 4 to 7 and 46 to 47. Most of the exonic rearrangements (deletions and duplications) were identified by panel P034, compared to the P035 panel. Also, a novel pathogenic single exon triplication was detected in the dystrophin gene.
