





1. Background
Since the description of very slowly propagating waves of “cortical spreading depression” in old studies of Leao in 1944 (1) and the following studies by Grafstein (2) describing the depolarizing nature of the waves, spreading depolarization (SD) has become more and more important in the pathophysiology of some neurological diseases such as epilepsy, stroke, traumatic brain injury and migraine with aura (3-5). The depolarization waves in SD propagate as concentric circles starting from a focus in the brain and affect many neurons and glial cells through the spreading way. Producing nearly complete prolonged depolarization for about 5 to 30 minutes in neurons, SD affects many molecular and cellular processes in neurons and astrocytes and changes the concentration of different ions and neurotransmitters in the tissue. It leads to impairment of neuronal electrical activity and finally transient, but long-lasting periods of nervous system dysfunction, hence naming “spreading depression” by Leao (5-7). In normal conditions, ion channels located within the cell membrane of neurons and astrocytes, function to keep the balance of the related ions between intra- and extracellular spaces. Finding appropriate ways to control SD, therefore, needs focusing on the behavior of these channels. The classic antidepressant, Nortriptyline, was chosen in this research to study some aspects of the spreading depolarization phenomenon mainly due to its inhibitory effects on voltage-gated (8), Ca2+-activated (8) and inwardly rectifying (Kir) potassium channels (9, 10) in addition to blocking the inactivated state of voltage-gated sodium channels (11, 12) and increasing intracellular calcium concentration (13). The drug also inhibits noradrenergic and serotonergic reuptake (14, 15), works as an NMDA receptor antagonist and inhibits the related Ca2+ influx (16), modulates endogenous opioid system (17) and finally blocks histaminergic (18) and muscarinic acetylcholine receptors (19).
2. Methods
2.1. Experiments
Adult male Wistar rats in the age range of 9 to 11 weeks were used. Animal care and experiments were in agreement with the National Institutes of Health guide for the care and use of laboratory animals. At the day of the experiment, each rat was anesthetized by isoflurane and immediately sacrificed, the brain was removed and put in ACSF-I solution (contained NaCl = 124, KCl = 4, CaCl2 = 1.0, NaH2PO4 = 1.24, MgSO4 = 1.3, NaHCO3 = 26, and glucose = 10 (all in mM)) at 4°C while continually receiving CO2/O2 with 5%/95% proportion. The brain was then placed in an automatic slicer device while drowned in ACSF-I solution and cut into transverse 500 µm-thick slices. The slices were then transferred to an ACSF-I pre-incubation chamber at 32°C. After 30 minutes, the solution was changed to ACSF-II by adding 1 µL/mL of 1M CaCl2 and after at least another 30 minutes of incubation, the slices were ready to be used in the electrophysiological recording device.
2.2. Groups
The slices were grouped randomly into control or nortriptyline groups. In the control group, recordings were performed pre, during, and post SD, while the slice was being washed with ACSF-II. In the nortriptyline group, recordings were done first during ACSF-II washing (pre SD). Then the washing solution was replaced by 100 µM nortriptyline solution, and after 30 minutes, SD was induced. The recordings were repeated during SD and after it (post SD) while nortriptyline washing was continued. Extra- and intracellular recordings were successful in 8 to 14 and 5 to 10 slices, respectively.
2.3. Electrophysiological Recording
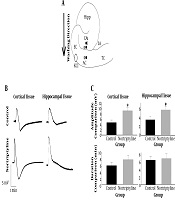
Each brain slice was put in the seat of an interface-type tissue slice super fusion system used for electrophysiological recordings (Brain/Tissue Slice Chamber System, Warner instruments) to get long-time access to the wet alive tissue. Four electrode tips were placed on each slice: one stimulating, one intracellular, and two extracellular electrodes. The intracellular electrode was a sharp glass micropipette filled with 2M K+-methyl sulfate with a resistance of 50 to 90 MΩ which its tip was put in the CA1 region of the hippocampus (Figure 1A). The reference electrode and the connection to the intracellular micropipette were symmetric Ag/AgCl bridges. Intracellular currents were injected within the range of 0.2 to 0.5 nA to see the irritability properties of hippocampal pyramidal neurons. The current pulses were passed via an active bridge circuit and bridge balance was adjusted during the recording time. Tip of one of the extracellular electrodes was placed in CA1 and the other in the auditory cortex, in the points indicated in Figure 1A. Each extracellular electrode had two plastic parts, one part filled with ACSF-II and the other with KCl, and was bridged by a ceramic part. A metal and a glass micropipette were connected to the KCl, and ACSF-II sides, respectively. The resistance of extracellular electrodes was 2 - 5 MΩ. Inhibitory postsynaptic potentials (IPSPs) were applied to the CA1 region of the hippocampus by putting the stimulating electrode on alveus of the hippocampus and activating antidromic post-synaptic activity in this region (20). The concentration of the nortriptyline solution was 100 µM, which is the concentration at which the drug can inhibit the function of many channels (8-16, 21). SD waves were induced by adding one small drop of 3 M KCl (the exact route of induction has been previously discussed (22). Figure 1A shows the point at which the KCl drop was added. The data measured in control and nortriptyline groups included: amplitude and duration of SD waves in hippocampal and cortical tissue spaces and the resulted changes in hippocampal intraneuronal spaces; resting membrane potential (RMP), primary depolarization amplitude, peak spike amplitude and number of induced spikes in hippocampal pyramidal cells in current injection sessions, pre and post SD; RMP, the amplitude and the duration of afterhyperpolarization waves in the pyramidal cells in IPSP sessions, pre and post SD. The data were collected and measured using Axoscope 12 software.
2.4. Data Analysis
Independent samples t-test or Mann-Whitney U tests were used to compare SD waves between nortriptyline and control groups and repeated measures analysis of variance (ANOVA) to compare current injections or IPSPs pre and post SD between the two groups. Also paired t-test was performed to compare cell membrane properties following current injection or IPSP in each of the control or nortriptyline groups. SPSS software v.25 was used for data analysis. The data were reported as mean +/- S.E.M or mean +/- S.D as is noted in each table or figure. Significance was considered as p values less than 0.05.
3. Results
3.1. Effect of Nortriptyline on SD Waves
Statistical analysis showed a significant difference in the amplitude of SD waves in both cortical and hippocampal tissues between nortriptyline and control groups (P value = 0.010 and 0.037, respectively). There was no significant difference in the duration of SD waves recorded in hippocampal or cortical tissues (P > 0.05). The pattern of SD waves in cortical and hippocampal tissues recorded by extracellular electrodes and the results of comparing the recorded values are depicted in Figure 1 and Table 1.
 and extracellular (E) electrodes (close circles) on a schematic representation of the hippocampus and surrounding neocortex. The point at which KCl drop was added is also shown with an open oval. I, intracellular; E, extracellular; Hipp, hippocampus; EC, entorhinal cortex; AC, auditory cortex; TC, temporal cortex; LA, lateral amygdala; B, shape of SD waves recorded in hippocampus and cortex by extracellular electrodes. Note the difference between shapes of the waves in hippocampus and cortex; C, the amplitude and duration of SD waves in hippocampal and cortical tissues were compared between nortriptyline and control groups. *, P < 0.05. Error bars are SEM.")
A, Positions of intra- (I) and extracellular (E) electrodes (close circles) on a schematic representation of the hippocampus and surrounding neocortex. The point at which KCl drop was added is also shown with an open oval. I, intracellular; E, extracellular; Hipp, hippocampus; EC, entorhinal cortex; AC, auditory cortex; TC, temporal cortex; LA, lateral amygdala; B, shape of SD waves recorded in hippocampus and cortex by extracellular electrodes. Note the difference between shapes of the waves in hippocampus and cortex; C, the amplitude and duration of SD waves in hippocampal and cortical tissues were compared between nortriptyline and control groups. *, P < 0.05. Error bars are SEM.
| Control (N = 10) | Nortriptyline (N = 7) | Significance (P < 0.05) | |
|---|---|---|---|
| SD amplitude, mV | |||
| Cortex | 5.08 ± 2.92 | 8.03 ± 3.48 | Yes |
| Hippocampus | 4.25 ± 2.41 | 6.14 ± 3.63 | Yes |
| SD duration | |||
| Cortex | 5.74 ± 3.59 | 6.64 ± 1.57 | No |
| Hippocampus | 6.38 ± 4.77 | 6.16 ± 1.55 | No |
Characteristics of Spreading Depolarization Waves. Results of Measuring Amplitude and Duration of SD Waves, Recorded by Cortical and Hippocampal Extracellular Electrodes in Nortriptyline and Control Groups Are Listeda
The pattern of SD waves in intracellular recordings is shown in Figure 2. No significant difference was seen in the amplitude or duration of SD waves recorded from hippocampal pyramidal neurons in the nortriptyline group vs. control after statistical analysis (P > 0.05).
3.2. Effect of Nortriptyline on Current Injection Properties
The difference in the change of current injection properties including early depolarization amplitude, the number of produced spikes, and peak spike amplitude due to SD were not significant between the nortriptyline and control groups. In addition, there was no significant difference in the referred properties between pre and post SD phases in each of the groups. Figure 2 shows the sample waves and Table 2 lists the resulted measurements.
 and IPSP (C) in hippocampal pyramidal cells pre and post SD.")
A, The shape of membrane potential changes in intracellular recording of hippocampal pyramidal neurons; B and C, shapes of current injection (B) and IPSP (C) in hippocampal pyramidal cells pre and post SD.
3.3. Effect of Nortriptyline on IPSP
The results showed that changes RMP and in the characteristics of IPSP following SD were not significantly different between nortriptyline and control groups, nor was any change after SD induction (post-SD) significant relative to pre SD phase in each of the control or nortriptyline groups (Figure 2 and Table 2).
| Current Injection | |||||
|---|---|---|---|---|---|
| Control (N = 5) | Nortriptyline (N = 5) | Significance (P < 0.05)c | |||
| Pre SD | 3 - 10 Min Post SD | Pre SD | 17 - 23 Min Post SDb | ||
| RMP before injection, mV | -62.90 ± 20.28 | -58.46 ± 15.25 | -48.53 ± 11.10 | -52.53 ± 21.17 | No |
| Applied current, nA | 0.27 ± 0.15 | 0.19 ± 0.13 | 0.54 ± 0.08 | 0.53 ± 0.10 | No |
| Depolarization, mV | 4.52 ± 2.96 | 3.96 ± 2.64 | 4.18 ± 2.34 | 4.27 ± 3.70 | No |
| Peak Amp., mV | 58.58 ± 15.58 | 46.10 ± 40.76 | 30.42 ± 11.07 | 6.67 ± -d | No |
| No of spikes, Mode | 1 | 1 | 1 | 0 | No |
| IPSD | |||||
| Control (N = 5) | Nortriptyline (N = 5) | Significance (P < 0.05)c | |||
| Pre SD | 5 - 10 Min Post SD | Pre SD | 5 - 10 Min Post SD | ||
| RMP before IPSP, mV | -54.53 ± 18.28 | -49.88 ± 17.75 | -59.23 ± 6.77 | -54.98 ± 14.14 | No |
| AHP Amp., mV | 1.55 ± 1.10 | 3.26 ± 4.50 (0.26 ± 1.68e) | 0.80 ± 0.45 | 0.11 ± 0.15 (-2.04 ± 1.09e) | No |
| AHP duration, ms | 334.57 ± 201.35 | 269.62 ± 219.30 | 320.50 ± 252.50 | 272.96 ± 273.55 | No |
Electrophysiological Properties of Hippocampal Pyramidal Neurons During Current Injection and IPSPa
4. Discussion
The primary aim of this study was to clarify some molecular aspects of spreading depolarization, as an important mechanism in the pathophysiology of some neurological diseases like stroke, epilepsy, migraine, and traumatic brain injury (3-5). The shape of SD in an extracellular recording is typically triphasic. First, a small positive deflection occurs due to severe depolarization of the cells, the part that does not always exist in the records. The second phase, which is the most characteristic feature of SD, is a negative wave of about 5 to 20 mV due to loss of the function in neuronal population in the region. Finally, the third phase is a positive-going wave of small amplitude with a duration of about 30 seconds (7, 23). Interpretation of the increase in the amplitude of SD waves in cortical and hippocampal extracellular spaces by using nortriptyline, found in this research, needs reviewing the proven effects of the drug on various types of ion channels situated on neuronal and astrocytic cell membranes. Among different effects of nortriptyline on ion channels (mentioned in the background part of this article), just increase in the intraneuronal concentration of calcium and rise in the extracellular potassium concentration ([K+]o) due to blocking the astrocytic potassium reuptake mechanism (blocking Kir4.1) can explain the observed increase in the amplitude. The increase in [K+]o is probably more important since it is the most important factor in inducing SD and many studies refer to it as the exclusive triggering cause for initiating SD (5, 24-26). When nortriptyline blocks Kir4.1 channels on astrocytic cell membrane, the key mechanism of the cortical tissue to fight extracellular K+ collection will be impaired and more neurons will be prone to depolarization as the result of increasing the positive charge near the outer face of their cell membrane. This helps to commit a higher number of neurons into the SD process, hence increasing the amplitude. The interesting story is that blockade of Kir4.1 channels by nortriptyline is dependent on voltage and [K+]o. Therefore, in SD situation, which both of these factors are notably increased, the drug would block the channels more efficiently (9). The amplitude values in the hippocampal tissue were lower relative to the cortex, which is probably due to the innate properties of the hippocampus and is in line with previous studies (27-29).
The rise in [K+]o, increasing intracellular Ca2+ concentration, or any other effects related to nortriptyline did not change the duration of SD waves, in its depolarization or repolarization/hyperpolarization parts. This, in addition to the loss of significant changes in the individual neuronal electrophysiological properties during SD propagation, current injection or inhibition of CA1 pyramidal neurons, again highlights the idea that nortriptyline effects on SD are mostly the result of the increase in the number of neurons involved in the process, rather than a change in the depolarization properties of each neuron. However, after finishing SD, all neurons in a field repolarize nearly at the same time, which obviously will not cause any change in the duration of the wave. Of course, clarifying the exact molecular events in intra- and extracellular spaces following nortriptyline use needs applying methods, like patch-clamp, that are able to study each ion’s behavior separately.
Revealing different aspects of spreading depolarization will make the basis for finding ways to control the phenomenon and prevent complications in the related neurological diseases like stroke, epilepsy, traumatic brain injury, and migraine with aura. This study just shed light upon some molecular aspects of the process and showed that changing the function of neuronal and astrocytic channels especially inwardly rectifying potassium channels could be a suggestion for changing SD properties. Lowering [K+]o using openers of astrocytic Kir4.1 channels might be the first propose to control SD, which determines its effectiveness requires further research.
