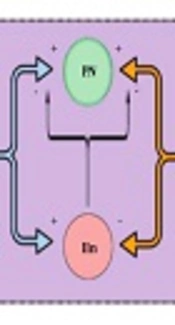
In 1965, Melzack and Wall (
18) published the gate control theory. Essentially, the gate, which represents the blockage of nociceptive signals from the spinal cord to the brain (and supraspinal nuclei) by nonnociceptive inputs and descending supraspinal control, is closed when nonnociceptive fibers stimulate an inhibitory interneuron in the DH. As inhibitory interneurons establish direct and indirect synaptic connections to projection neurons, their stimulation decreases the outputs for higher control and diminishes nociceptive transmission and consequent pain. On the other hand, when inhibitory interneurons are inhibited by nociceptive afferent inputs, the gate is opened, facilitating the transmission of nociceptive signals (
Figure 2) (
14,
18).
Disinhibition, in turn, implies a dysfunction of inhibitory interneurons, with a reduction in GABAergic or glycinergic control (
19). By analyzing the importance of these cells in the inhibition of pain, it is easy to understand that impaired activity of these interneurons can result in enhanced pain. Evidence suggests that disinhibition may be one of the most important underlying NP mechanisms, including pain relief signals by GABA administration and pain-like behaviors in animal models when intrathecal antagonists of GABA
A or glycinergic receptors are intrathecally administered (
5,
20).
The mechanisms suggested for disinhibition include death of inhibitory interneurons (1), reduced afferent drive towards inhibitory interneurons (2), depletion of GABA, alteration of GABA function when released (3), changes in inhibitory interneuronal membrane properties, and specific glycine alterations (4). In the next section, a brief discussion on each of these mechanisms is presented.
6.1. Loss of Inhibitory Interneurons and Reduced Afferent Drive
Moore et al. (
6) used 3 main peripheral nerve injury models to determine the inhibitory mechanisms. For identifying whether a reduction in inhibitory inputs occurs after injury and whether it happens pre- or postsynaptically, they measured the primary afferent-evoked inhibitory postsynaptic currents (IPSCs) in lamina II neurons, which occur due to GABA/glycine activity on postsynaptic receptors. They were found to be reduced, whereas the excitatory ones remained similar to the preinjury stage in 2 models. This finding supports the hypothesis of diminished inhibition following injury (
6).
They also observed a reduction in glutamate decarboxylase (GAD65), a GABA synthesizing enzyme, suggesting GAD65 downregulation ipsilateral to the injury. In addition, they observed that GAD65 returned to its baseline level within 4 weeks after injury, at least in the CCI model, which supports downregulation over permanent loss of enzyme. On the other hand, they examined apoptotic neurons, as neuronal death might explain the reduction of GAD65; however, it could not accurately explain the normalization of enzyme levels after some weeks.
Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) was used to identify apoptotic cells (
6). The TUNEL-positive cells were identified at 1 week after injury in the superficial DH. However, many did not present the neuronal NeuN marker (
6), necessitating further studies to determine whether these cells are neurons, and if so, whether they are GABAergic.
Scholz et al. (
21) supported the occurrence of apoptosis in the superficial DH through observation of TUNEL-positive nuclei in CCI, SNL, and SNI models. In addition, they combined caspase-3 and NeuN labelling to identify apoptotic neurons. Through stereological analysis of NeuN-positive cells, they found a 22% reduction in neurons at 4 weeks after injury, consistent with previous findings (
22). By using GAD67 as a marker for GABAergic interneurons, they observed a reduction of about 24% in these cells.
A series of experiments on this subject was developed because of the uncertainty about cell markers. In the SNI model, Polgar et al. (
23) examined neuronal density in the superficial DH with a stereological method and found no significant reduction, neither in the first nor in the fourth week after injury. Another study also showed similar results, using the CCI model (
24). In addition, they analyzed caspase-3 immunoreactivity in this region, indicating a moderate to high level of immunoreactivity to the injury on both sides (contralateral and ipsilateral), with no morphological alterations in the nucleus or NeuN positivity in the cells (
23).
To examine whether these cells could be astrocytes, they analyzed the relationship of strong caspase-3 positivity to antibody labelling against glial fibrillary acidic protein (GFAP), as an astrocyte marker. A great majority of the cells were labelled by both markers, suggesting that they could be astrocytes. Moreover, as all astrocytes were virtually marked with caspase-3, this marker is unlikely to be related to apoptotic signals in these cells (
23).
TUNEL staining was also examined, presenting some interesting features. The positive cells were distributed both in the grey and white matters, presenting no NeuN positivity. Iba-1 immunoreactivity, a marker of microglia, was also found in these cells. This finding, along with the fact that some cells were distant in the white matter, suggested that these cells could be microglia (
23).
Another characteristic was the proximity of neurons to TUNEL-positive cells, sometimes even overlapping each other, which could possibly explain the combined labelling of these markers in previous studies, although these cells were always distinguishable by the nucleus morphology. These findings altogether suggest that in many studies, apoptotic cells are likely to be glial cells, not neurons (
23). Furthermore, no specific GABA- or glycine-like immunoreactive neuron reduction was reported in the CCI model (
25).
Some discrepancies in the findings may be explained by methodological differences and variations in data analysis, whereas some are not yet well understood. For instance, different conclusions about TUNEL-positive cells depend on whether these cells are considered neurons or astrocytes, based on the cell morphology and markers. According to this classification, conclusions change considerably.
To examine whether there is a significant reduction in GABA explaining disinhibition, Polgar and Todd (
26) analyzed the level of GABA neurotransmitters in GABAergic terminals through immunogold labelling for electron microscopy among SNI rats and considered this approach as optimal for this purpose. They found no evidence of significant loss in GABAergic boutons or depletion of GABA
A receptors. This finding was unexpected, as any level of GABA depletion was speculated to support the disinhibition hypothesis. Nevertheless, they suggested that reduced excitatory afferent drive to GABAergic interneurons may account for or at least contribute to abnormal GABA activity, leading to disinhibition.
Reduced excitation of these interneurons may occur through a variety of mechanisms, including impaired excitatory drive from primary afferents to these cells (
4,
5). In this regard, Kohno et al. (
27) suggested that there is a reorganization of excitatory inputs and a loss of most monosynaptic inputs to lamina I neurons, based on the analysis of excitatory postsynaptic currents (EPSCs), evoked by primary afferents in this layer.
Recently, Leitner et al. (
28) explored the mentioned hypothesis in mice expressing green fluorescent protein (GFP) under the control of GAD67 promoter. These mice are very useful, as GFP is expressed in subsets of inhibitory interneurons in specific mouse lines. They identified a reduction in excitatory currents, directed to GFP cells after CCI, consistent with the disinhibition hypothesis. In addition, the dorsal root was stimulated, and the evoked responses from Aδ and C fibers were higher than the controls, suggesting no loss of synapses, accompanied by the decreased probability of neurotransmitter release to the synaptic cleft.
Also, activation of the neuronal activity marker, c-Fos, under noxious stimulation was lower in GABAergic neurons in lamina II after CCI, compared to the control group. Reduced activation of these inhibitory interneurons was reported, even with nociceptive inputs. No changes occurred in the density or morphology of dendritic spines, which presents evidence against the loss of excitatory synapses. Todd (
5), however, noted the difficulty of interpreting these results, as the dendritic spines are likely to be only part of excitatory synaptic inputs to lamina II, as other types of synapses are established and many cells have few dendrites (
5).
In this regard, neuronal death has been thoroughly discussed in scientific circles and many researchers do not consider it as a major possibility, based on adequate scientific evidence. Nevertheless, reduced primary afferent drive is very likely to contribute to disinhibition, even though the degree of this contribution has not been yet established.
6.2. Changes in GABA Synthesis or Function
Ibuki et al. (
29) analyzed GABA immunoreactivity in several intervals after CCI. They observed an important reduction in the GABA profile; in fact, no profile could be seen in 2 weeks, while within 7 weeks, partial recovery occurred. Eaton et al. (
30) reported similar results on the GABA immunoreactivity profile. However, recent studies, including those by Polgar and Todd (
26), have not observed a reduction in GABAergic axons, suggesting the possible downregulation of GAD65; however, it was inadequate to disrupt the GABA level on the terminals (i.e., GABA synthesis).
In addition, other studies have reported no evidence of GABAa reduction (
6,
25,
28). The discrepancy between these studies and those by Ibuki et al. and Eaton et al. may be related to technical considerations, such as differences in cytological preparation and fixation. One example of unexplained data is the loss of GABA in both ipsilateral and contralateral sides to the injury, as shown by Ibuki et al. and Eaton et al., even with no signals of NP in the contralateral side.
Other mechanisms of GABA changes resulting in disinhibition include changes in the neurotransmitter function. One of the most interesting examples is the GABA shift of function, resulting in depolarization instead of hyperpolarization of postsynaptic cells as projection neurons (
31,
32). This shift changes the direction of chloride (Cl
-) flux, which is determined by the ratio of Cl
- equilibrium potential (E
Cl) to the resting membrane potential of the cells (
1).
Generally, E
Cl is more negative than the resting membrane potential, because of the continuous removal of Cl
- from the cell. This explains the normal activity of GABA in these terminals, which leads to Cl
- influx and subsequent hyperpolarization of the cell. When E
Cl is altered, being less negative than the resting membrane potential, GABAa opening results in Cl
- efflux and causes abnormal cell depolarization (
31,
32). Changes in E
Cl may happen due to changes in Cl
- cotransporters, probably triggered by the brain-derived neurotrophic factor (BDNF) stimulation of tropomyosin receptor kinase B (TrkB) (
1,
31,
32).
Since BDNF is released by activated microglia, intrathecal injections of ATP-activated microglia produce behavioral changes, resembling those in NP, whereas inactivated microglia have no such effects (
31). Likewise, the subsequent blockage of BDNF signaling to the TrkB receptor prevented allodynia (
31). Therefore, microglia may be of great importance as an underlying mechanism of NP; nevertheless, alterations in glial-neuronal interactions in NP are not restricted to interneurons. Therefore, not only depletion of GABA, but also its functional changes are being analyzed as possible mechanisms of disinhibition. The latter hypotheses, especially those involving the Cl
- flux inversion hypothesis, are promising due to lack of evidence on GABA depletion.
6.3. Changes in Inhibitory Interneuronal Membrane Properties
Another hypothesis suggests that intrinsic plasticity may be disinhibitory (changes in neuronal membrane excitability including inhibitory interneuron membranes). These modifications in electrical membrane properties may include presynaptic changes in the curve of action potential, changes in firing patterns, and reduced excitability (
1).
Schoffnegger et al. (
33) conducted a study regarding the physiological properties of lamina II GABAergic neurons after nerve injury. They used GFP-expressing mice under the control of GAD67 promoter. They found that the membrane excitability of these neurons, synaptic inputs, and firing pattern had no significant differences, compared to the sham animals. Therefore, this study does not support the changes in active or passive membrane properties in spinal GABAergic interneurons following nerve injury. However, it should be noted that only one-third of all GABAergic interneurons were labelled with this method, although it may represent a proper sample of these neurons.
As discussed by Sandkuhler (
1),
plateau potentials instead of tonic firing provoke an amplified response to nociceptive inputs, as it is more intense with prolonged postdischarge periods. Activation of GABA
A or GABA
B receptors inhibits the
plateau potentials and converts them to the firing pattern. Based on these findings, the firing pattern of inhibitory interneurons could be altered in NP. However, Schoffnegger et al. (
33) found no supporting evidence for this hypothesis.
Although this hypothesis found no major support regarding interneurons, alterations in ionic voltage-gated channels are well established as a possible mechanism of NP, related to nociceptors and projection neurons (
1). Increased excitability of the membrane of deep DH neurons has been observed due to a shift in the steady-state properties of sodium channels (
34). Likewise, upregulation of voltage-gated sodium channels of Na(v)1.3 subtype ipsilateral to the injury was found in neurons coexpressing neurokinin 1 receptor (NK1R) markers, thereby suggesting them as projection neurons (
35).
In addition, there is clinical evidence of pain relief with the use of sodium-channel blockers in NP, such as lidocaine, commonly used in form of topical patches (
36,
37). Although evidence on its efficacy remains conflicting (
38), due to lack of high-quality randomized controlled studies, there is substantial evidence on its efficacy and safety in clinical settings and individual studies, especially regarding localized NP (
36,
39).
6.4. Specific Glycine Alterations
Glycine is thought to be used by some lamina I-III interneurons as a cotransmitter. Some studies showed that neurons normally use one of these neurotransmitters, despite expressing both (
5). In addition, allodynia was observed when blocking glycine receptors (
40). Lu et al. (
41) proposed that NP is related to the glycinergic inhibition of excitatory interneurons, expressing the γ-isoform of protein kinase C (PKCy) on lamina IIi (inner layer). The PKCy interneurons are activated by myelinated, low-threshold mechanoreceptors (Aβ).
Both glycinergic interneurons and Aβ fibers converge inputs by PKCy interneurons. Therefore, glycinergic neurons from lamina III decrease Aβ synapse effects on PKCy interneurons, inhibiting these excitatory interneurons as a gate control. Following injury, this inhibition may be partially lost, allowing a greater transmission of excitatory inputs leading to NP. However, these findings were observed in the L5 segment, but not in the upper segments through which the nociceptive signal necessarily flows before reaching L5; this finding raised some doubts about this hypothesis.
Foster et al. (
42) also found evidence consistent with the strong innervation of spinal glycinergic interneurons by nonnociceptive sensory neurons. In addition, ablation of glycinergic neurons resulted in hypersensitivity and spontaneous aversive behaviors, whereas their exogenous activation ameliorated neuropathic hyperalgesia (
41).


")
")
