



1. Background
Wilson’s disease (WD) is an autosomal recessive genetic disorder caused by mutations in the ATP7B gene on chromosome 13, leading to defective copper metabolism. Over 700 mutations associated with this disease have been identified, and most patients are heterozygous carriers (1). Wilson’s disease can present at any age, although most patients are diagnosed between the ages of 5 and 35 (2). The global prevalence of WD is estimated to be one case per 30,000 individuals, with a higher prevalence in regions where consanguineous marriages are more common (3). This disease has a wide range of clinical manifestations, making its diagnosis challenging in a clinical setting. Wilson’s disease can affect the liver, nervous system, psychiatric system, eyes, hematological system, musculoskeletal system, heart, kidneys, skin, and other organs (4). It should be considered in the differential diagnosis of any hepatic presentation (2).
The neurological manifestations of WD include a broad spectrum of movement disorders such as tremor, bradykinesia, rigidity, dystonia, chorea, dysarthria, dysphagia, and less common neurologic symptoms. These neurologic signs typically emerge in the second and third decades of life, usually approximately a decade after the onset of hepatic involvement (5-7). Ocular manifestations, such as Kayser-Fleischer rings and sunflower cataracts, result from copper accumulation in these organs. Kayser-Fleischer rings are observed in nearly all patients with neurologic presentations and in 50% of those with hepatic presentations (2, 5, 8).
The diagnosis of WD is based on the Leipzig scoring system (6), and neurologic symptoms are assessed via the Unified Wilson’s Disease Rating Scale (UWDRS) and the Global Assessment Scale (GAS) (9, 10). These systems help predict the severity of neurologic symptoms and assist in patient management (5, 10). The presence of Kayser-Fleischer rings and low serum ceruloplasmin levels are typically sufficient for diagnosis. In their absence, a comprehensive evaluation of copper metabolism is needed (2).
Treatment options for WD include D-penicillamine, trientine, zinc, tetrathiomolybdate, and dimercaprol. Once diagnosed, these medications are administered for life. Liver transplantation is reserved for patients with acute liver failure and decompensated cirrhosis (2). Early diagnosis and treatment of WD generally lead to patient improvement, whereas delayed diagnosis and disease progression can result in irreversible complications (5). Given the prevalence and broad range of neurologic symptoms in this disease, early recognition and correlation with other organ involvement and clinical tests are crucial.
2. Objectives
Despite advances in understanding WD, the heterogeneity and delayed recognition of its neurologic manifestations remain significant clinical challenges. The present study aims to systematically characterize the spectrum and progression of neurologic symptoms in WD in the Iranian population. We seek to improve early diagnosis, reduce morbidity and mortality, and optimize therapeutic strategies for this treatable disorder.
3. Methods
3.1. Study Design
All patients diagnosed with WD who presented at Rasoul Akram Hospital between 2002 and 2022 were included in this study. The inclusion criteria were a diagnosis of WD based on the Leipzig scoring system (6) with confirmation by a movement specialist and the absence of other diagnoses. Patients with only gastrointestinal symptoms and incomplete records were excluded.
3.2. Primary Outcome
The present study primarily aimed to assess the neurological findings in patients diagnosed with WD who were referred to Rasoul Akram Hospital. This includes evaluating motor function using the UWDRS, cognitive ability using the mini-mental state examination (MMSE), and psychiatric symptoms using the diagnostic and statistical manual of mental disorders, fifth edition (DSM-5). These examinations were performed by board-certified neurologists blinded to laboratory and imaging results. Outcomes were categorized as the presence or absence of symptoms and functional disabilities.
3.3. Data Collection
Clinical symptoms and laboratory data were obtained from hospital records. Imaging features included abdominopelvic sonography and brain MRI, extracted from radiologic reports generated by board-certified radiologists blinded to clinical data. Brain MRI was performed using T1-weighted, T2-weighted, and FLAIR sequences, with any unusual signal changes documented.
3.4. Sample Size
This study used a census sampling method to include all patients who presented to Rasoul Akram Hospital between 2002 and 2022.
3.5. Data Analysis
Data analysis was performed using SPSS version 26 (IBM Corp.). Descriptive statistics included frequencies and percentages for categorical variables and mean ± standard deviation (or median and interquartile range) for continuous variables. Results were summarized in tables and figures to illustrate patterns in neurologic manifestations.
4. Results
4.1. Study Population
In the present study, the medical records of patients diagnosed with WD who were referred to Rasoul Akram Hospital, Tehran, Iran, from 2002 to 2022 were reviewed. During this period, 116 records with an initial diagnosis of WD were available. Among these patients, 25 presented with only gastrointestinal symptoms and were excluded from the study as specialized neurological examinations were not conducted. Twelve patients were eventually diagnosed with conditions other than WD. Eleven patients had multiple hospital visits and were evaluated over several time periods. Eight patients left the hospital before completing their evaluations, resulting in incomplete records. Consequently, a total of 56 cases were excluded from the study. The analyses were conducted on 60 confirmed cases of WD with neurological manifestations. Among the 60 patients, 27 were women (45%) and 33 were men (55%), with a mean age of 20.78 ± 12.00 years.
4.2. Laboratory Findings
The laboratory analysis revealed that the hemoglobin (Hb) level in males averaged 12.9 ± 2.1 g/dL, whereas in females, the average Hb level was 11.5 ± 1.9 g/dL. Anemia appeared in 16 males (76.2%) and 8 females (34.8%). The platelet counts varied widely, averaging 107.5 ± 87.1 × 103/µL among the 44 patients. Twenty-six patients (59.1%) had thrombocytopenia. Additionally, the white blood cell (WBC) count was noted to be 4.9 ± 1.72 × 103/µL among 43 patients (Table 1). The liver enzyme and total bilirubin levels of the patients in this study are shown in Table 2. Among the patients, 16 (42.1%) showed elevated alanine aminotransferase (ALT), 33 (86.8%) had increased aspartate aminotransferase (AST), 27 (84.3%) exhibited elevated alkaline phosphatase (ALKP), and 4 (22.2%) had elevated total bilirubin levels (Table 1).
| Variables | Mean ± SD (Range) | No. (%) |
|---|---|---|
| WBC (n = 43) | 4.9 ± 1.72 (1.9 - 9.2) | - |
| Hb | ||
| Male (n = 21) | 12.9 ± 2.1 (9.1 - 17.7) | Anemia: 16 (76.2) |
| Female (n = 23) | 11.5 ± 1.9 (9.3 - 15) | Anemia: 8 (34.8) |
| Platelet (n = 44) | 107.5 ± 87.1 (22 - 402) | Thrombocytopenia: 26 (59.1) |
| ESR (n = 18) | 10.33 ± 6.4 (1 - 24) | - |
| ALT (n = 38) | 41.73 ± 61.88 (7 - 390) | High: 16 (42.1); normal: 22 (57.9) |
| AST (n = 38) | 34.5 ± 26.17 (17 - 150) | High: 33 (86.8); normal: 5 (13.2) |
| ALKP (n = 32) | 244 ± 215.15 (122 - 862) | High: 27 (84.3); normal: 5 (15.7) |
| Total bilirubin (n = 18) | 0.9 ± 0.911 (0.4 - 3.4) | High: 4 (22.2); normal: 14 (77.8) |
Abbreviations: WBC, white blood cell; Hb, hemoglobin; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ALKP, alkaline phosphatase.
| Clinical Features | Total Number | Positive | Prevalence Percent |
|---|---|---|---|
| Dystonia a | 40 | 34 | 85 |
| Tremor b | 34 | 27 | 79.4 |
| Dysarthria | 44 | 30 | 68.2 |
| Gait disturbances c | 40 | 26 | 65 |
| Hypobradykinesia | 42 | 22 | 52.4 |
| Neuropsychological disorders d | 30 | 14 | 46.7 |
| Rigidity | 43 | 19 | 44.2 |
| Increased DTR | 32 | 14 | 43.8 |
| Dysphagia | 36 | 12 | 33.3 |
| Ataxia | 15 | 5 | 33.3 |
| Increased muscle tone | 24 | 7 | 29.2 |
| Drooling | 33 | 8 | 24.2 |
| Upward plantar reflex | 34 | 5 | 14.7 |
| Sensory disorder | 24 | 3 | 12.5 |
| Seizure | 30 | 3 | 10 |
| Chorea-athetosis | 21 | 2 | 9.5 |
a Dystonia: Generalized (23.5%), dystonia in four limbs (14.7%), dystonia with tremor (14.7%), upper limb, speech, facial, and unspecified types.
b Tremor: At rest (26.5%), action (29.4%), postural (14.7%).
c Gait disturbances: Dystonic (26.9%), ataxic (15.3%), spastic (11.5%), wide-based, atonic, inability to walk, and unspecified types.
d Neuropsychological disorders: Depression (4 patients), hallucinations (2 patients), insomnia (2 patients), developmental delay (2 patients), low MMSE scores (4 patients), cognitive impairment, irritability, and delusions (1 patient).
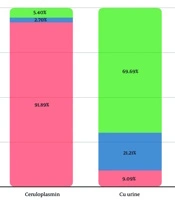
The copper profile of patients is shown in Figure 1. We did not have any data regarding the initiation of drug therapy; therefore, the observed values may reflect both untreated disease progression and potential variability due to incomplete treatment histories (Figure 1).

Copper profile of patients
4.3. Gastrointestinal Findings
Gastrointestinal involvement was common, with liver cirrhosis observed in 67% of patients and splenomegaly in 36%. Further details on hepatosplenomegaly and cirrhosis, including ultrasound findings and subgroup distributions, are provided in Figure 2.

Liver cirrhosis and hepatosplenomegaly based on abdominopelvic sonography of patients
4.4. Neurologic Findings
Neurologic involvement was prominent in the study population, with dystonia (85%), tremor (79.4%), and dysarthria (68.2%) as the most frequent manifestations (Table 2). Gait disturbances (65%) and hypobradykinesia (52.4%) were also common, while neuropsychological disorders (46.7%) and rigidity (44.2%) occurred less frequently. Increased deep tendon reflexes (43.8%) and rare signs like chorea-athetosis (9.5%) were also observed, highlighting the diverse neurologic presentation of WD in this population. A detailed breakdown of neurologic findings, including subtype distributions (e.g., tremor types, dystonia patterns), is provided in Table 2.
Ophthalmologic examinations with a slit lamp were conducted on 50 patients. Among them, 36 patients tested positive for Kayser-Fleischer rings, whereas 14 patients (28%) tested negative. The negative results were predominantly from patients who were already receiving treatment for WD, which likely accounts for these outcomes.
4.5. Imaging Findings
Brain MRI, a key modality for detecting neurologic involvement in WD, revealed characteristic abnormalities in our patients. Radiologic reports, utilizing standard T1-weighted, T2-weighted, and FLAIR sequences, documented signal changes in the putamen, midbrain, thalamus, and other regions, as illustrated in Figure 3. The pathognomonic "face of the giant panda" sign was identified in 18.75% of patients (Figure 3).

Signal changes in the brain MR images of patients
5. Discussion
We analyzed 60 confirmed cases of WD with neurological manifestations from 116 records at Rasoul Akram Hospital (2002 - 2022). Neurological symptoms were present in 64.5% of WD patients, aligning with Lankarani et al. (56%) (11) but exceeding Asadi Pooya et al.’s Shiraz cohort (12). The mean age of onset was 20.78 years, consistent with typical WD onset (2), though Soltanzadeh et al. reported an earlier onset of 16 years (13). Notably, three patients exhibited symptoms before age 6, suggesting earlier neurological involvement than previously documented (2).
5.1. Neurologic Findings
Our study exclusively focused on neurological WD patients, identifying dystonia (85%), tremor (79.4%), dysarthria (68.2%), and gait disturbances (65%) as the most frequent neurologic manifestations. These findings are largely consistent with those reported in previous studies (Table 3). The differences in percentages may be due to the inclusion of a larger patient population, including WD patients with gastrointestinal symptoms. As seen in Table 3, the percentages in Asadi Pooya and Malekzadeh’s studies are much lower than others, likely because they included gastrointestinal cases in their population (11, 12). A summary of findings from prior studies can be found in Table 3.
| Study | Study Design | Location | Year Conducted | Mean Age | Most Common Neurologic Manifestations (%) | Imaging Findings (MRI) (%) |
|---|---|---|---|---|---|---|
| Present study | Case series | Rasoul Akram Hospital, Tehran, Iran | 2002 - 2022 | 20.78 | Dystonia (85); tremor (79.4); dysarthria (68.2); gait disturbances (65); hypobradykinesia (52.4); neuropsychological disorders (46.7) | Signal changes in: Putamen (45.8); midbrain (31.2); thalamus (27.1); pons (25); caudate (18.7); tegmentum (16.7); panda sign (18.7) |
| Lankarani et al. (11) | Case-control | Shiraz Organ Transplantation Center, Shiraz, Iran | 2000 - 2014 | 16 a, 22 b | Tremor (40.5); ataxia (32.4); depression (24.3); dysarthria (21.6); drooling (21.6); fine motor task (17.6); gait disorder (17.6); anxiety (16.4); neurosis (16.2); seizure (16.2) | |
| Asadi Pooya et al. (12) | Case series | Namazee Hospital, Shiraz, Iran | 1990 - 2004 | 11 ± 7 | Dystonia (15.3); dysarthria (12.6); tremor (11.7); gait disturbances (9); impaired school performance c (7.2); psychosis c (7.2); other psychological symptoms c (9.9) | |
| Soltanzadeh et al. (13) | Case series | Shariati University Hospital, Tehran, Iran | 1984 - 2004 | 16 | Dysarthria (80); drooling (48); abnormal gait (44); psychiatric symptoms (44); tremor (44); dystonia in limbs (42); dystonia in face (36); dysphagia (30) | |
| Machado et al. (14) | Case series | University of Sao Paulo, Sao Paulo, Brazil | 1963 - 2004 | 19.6 | Dysarthria (91); gait abnormalities (75); risus sardonicus (72); dystonia (69); psychiatric sumptoms (67); rigidity (66); tremor (60); bradykinesia (58); dysphagia (50); postural instability (49) | |
| Lorincz (15) | Systematic review | - | 2010 | Around 17 | Dysarthria (85 - 97); dystonia (11 - 65); tremor (22 - 55); parkinsonism (19 - 62); ataxia (30); psychiatric features (30 - 50) | High T2 signals in: Lentiform and caudate n., thalamus, brain stem, white matter |
| Pfeiffer (16) | Systematic review | - | 2016 | 20 | Dysarthria in the vast majority; tremor (50); dystonia (69); parkinsonism (40); dysphagia (50); gait disturbances (45 - 75); cerebellar dysfunction (30); psychiatric manifestations (30 - 40) | High T2 and low T1 signal changes in: Basal, ganglia, brain stem, thalamus |
| Mulligan and Bronstein (17) | Systematic review | - | 2020 | 10 - 20 | Dysarthria (46 - 97); gait abnormality/ataxia/cerebellar (28 - 75); dystonia (38 - 69); parkinsonism (12 - 58); postural tremor (55); dysphagia (50) | Signal changes in: Basal, ganglia, thalami, pons, white matter, face of giant Panda (14) |
| Czlonkowska et al. (5) | Systematic review | - | 2017 | 20 - 30 | Tremor up to (55); dystonia (11 - 65) d, (38 - 65) e; parkinsonism (19 - 62); ataxia (30); chorea (6 - 16); dysarthria up tp (97); dysphagia (50); drooling (46); gait and posture (44 - 75) | Hyper or mixed intensive changes in T2-weighted images localized in: Putamina, globi pallidi, caudate n., thalami, pons |
| Dusek et al. (18) | Systematic review | - | 2019 | 20 - 40 | Dysarthria (100); dysphagia (50); drooling (70); gait/posture disturbances (44 - 75); dystonia (11 - 65); parkinsonism (19 - 62); ataxia (30) | Symmetric hyperintensities in t2-w images in: Putamen (45 - 85); caudate (30 - 60); anterolateral thalamic n. (30 - 60); pons (10 - 80); mesencephalon (20 - 70) |
| Salari et al. (19) | Systematic review | - | 2018 | Variable | Variable in different studies | Symmetric hyperintensities in T2-W in: Pons, midbrain, basal ganglia |
a Full hepatic failure patients (acute Wilson’s disease).
b Chronic Wilson’s disease.
c Classified as psychological sign and symptoms.
d As an initial symptom.
e During the course of disease.
5.2. Imaging Findings
In other studies, the MR images of patients have rarely been examined. In one study, the observed MRI changes included brain atrophy and increased signals in the putamen, globus pallidus, caudate nucleus, thalamus, and pons on T2-weighted images (5, 20, 21). The midbrain, cerebellum, corticospinal tract, and other white matter regions can also be affected (2, 5, 22). The characteristic finding on MRI in WD patients is the presence of the panda sign (5, 23) (Table 3). In a study involving MRI imaging of 22 patients, 19 patients (86%) had abnormal findings. Despite differences in absolute percentage values between this study and our study, the areas affected by WD are largely the same, indicating consistency between the two studies (5, 21). According to the 2018 study by Salari et al., specific MRI abnormalities in WD closely correlate with clinical signs and symptoms. Dysarthria and tremor are consistently associated with lesions in the globus pallidus and thalamus; chorea correlates with caudate nucleus lesions; nystagmus is linked to abnormalities in the lentiform nucleus and thalamus; rigidity corresponds to lesions in the lentiform nucleus, white matter, and thalamus; and dystonia is associated with changes in the globus pallidus and putamen (19). Unfortunately, further clinical research is necessary to better understand the diagnostic and prognostic implications of MRI findings in WD (5, 19).
5.3. Laboratory and Gastrointestinal Findings
In our study, 91% of patients exhibited low serum ceruloplasmin, aligning with WD hallmarks, though reduced levels may also occur in heterozygotes, nephrotic syndrome, or liver disease, while elevated levels can arise during acute-phase reactions, pregnancy, or oral contraceptive use. Elevated free serum copper (89%) directly reflected systemic copper overload, yet its measurement remains limited in practice. While 70% showed increased 24-hour urinary copper, 21% had normal values, likely due to collection challenges or early-stage disease. These findings emphasize the non-specificity of isolated biomarkers and reinforce the necessity of integrating clinical, genetic, and adjunctive tests (e.g., penicillamine challenge) for definitive WD diagnosis (17).
Hepatic involvement was prominent in our cohort, with 86.6% elevated AST, 84% elevated ALKP, and 67% liver cirrhosis, aligning with the progressive hepatic spectrum of WD from transaminase elevation to cirrhosis. Splenomegaly (34%) and elevated ALT (42%) further underscored chronic liver pathology, consistent with literature highlighting ascites, hepatosplenomegaly, and jaundice as common early hepatic presentations. While acute fulminant failure was rare, the high cirrhosis prevalence mirrors WD’s insidious progression, emphasizing the need for early diagnosis via biomarkers (e.g., AST/ALT ratio > 2.2) and copper studies to mitigate morbidity. Our findings reinforce hepatic WD’s heterogeneity and its critical role in timely intervention (16, 17, 24).
The observed anemia (76.2% males, 34.8% females) and thrombocytopenia (59.1%) align with the hematologic complications of WD, which are often secondary to chronic liver disease, hypersplenism, or copper-induced hemolysis (4, 24). The gender disparity in anemia prevalence may reflect iron deficiency or hormonal influences in females, though further investigation is warranted. Notably, the low platelet count (107.5 ± 87.1 × 10³/µL) and mild leukopenia (WBC 4.9 ± 1.72 × 10³/µL) are consistent with portal hypertension and splenic sequestration in advanced hepatic WD (4, 24). These hematologic abnormalities, while non-specific, reinforce the systemic nature of WD and highlight the need to integrate them with clinical and biochemical markers (e.g., ceruloplasmin, urinary copper) for comprehensive disease assessment.
5.4. Study Limitations
This single-center, retrospective study at Rasoul Akram Hospital has limitations, including a small sample size, lack of follow-up data, incomplete neurological assessments (particularly in gastrointestinal cases), and potential data loss over its long timeframe. Despite these constraints, the findings provide valuable insights for early diagnosis and management of WD in Iran, potentially reducing associated morbidity and mortality.
5.5. Conclusions
In patients approximately 20 years of age, the presence of neurologic symptoms such as dystonia, tremor, dysarthria, gait disturbances, and hypokinesia should raise clinical suspicion for WD, prompting further paraclinical evaluations for early diagnosis. The findings of this study can aid in the early diagnosis of WD in the Iranian population, potentially reducing the morbidity and mortality associated with this condition.
