



1. Introduction
Schaaf-Yang syndrome (SYS), with only 250 reported cases worldwide as of May 2022, is an autosomal dominant multisystem disease first identified by Dr. Schaaf in 2013 as a genetic variant of Prader-Willi syndrome (PWS) (1). The syndrome’s most prominent features include hypotonia, joint contractures, autism spectrum disorders, and intellectual disability or developmental delay (1). Schaaf-Yang syndrome is caused by pathogenic mutations in the maternally imprinted, paternally expressed melanoma antigen L2 (MAGEL2) gene (OMIM 605283), located on chromosome 15 in the Prader-Willi critical region (15q11 - 15q13). This gene is part of a cluster of imprinted genes essential for normal neurodevelopment; mutations on the paternal allele are harmful due to maternal imprinting (2, 3).
Schaaf-Yang syndrome is characterized by a broad range of clinical symptoms, including feeding difficulties, joint contractures, intellectual disability, developmental delay, infantile hypotonia, and autism spectrum disorder (ASD) (3). Early symptoms often include hypotonia and poor sucking, which indicate severe feeding difficulties in infancy. All individuals with SYS experience developmental delay, intellectual disability, and poor sucking during infancy (4). Affected individuals commonly exhibit significant delays in achieving motor milestones and language development, with developmental delay and intellectual disability being nearly universal (1, 2).
Respiratory distress is another critical symptom of SYS, potentially caused by respiratory muscle impairment due to muscular hypotonia or abnormalities in the brain’s respiratory center (5). Endocrine disorders, including hypogonadism, hypothyroidism, and growth hormone (GH) deficiency, are also prevalent, reflecting the syndrome’s extensive impact on multiple physiological systems (6).
Both SYS and PWS impair thermoregulation, possibly due to disruptions in oxytocin pathways (7). The MAGEL2 gene plays a significant role as a mammalian-specific regulator of hypothalamic neuroendocrine function, making the hypothalamus a key regulator of both physiological homeostasis and behavior. Growth abnormalities, sleep disturbances, and feeding difficulties are among the symptoms SYS patients may exhibit due to MAGEL2 dysregulation, which can result in altered levels of various hormones and neurotransmitters (8).
Recent genetic studies indicate that SYS is primarily caused by truncating mutations in the MAGEL2 gene, which disrupt critical pathways for cellular functions and protein recycling and have been associated with various neurodegenerative disorders. Due to clinical overlap with more common conditions like PWS, diagnosing SYS can be challenging and requires comprehensive genetic testing for confirmation (9). The MAGEL2 gene, part of the MAGE family of ubiquitin ligase regulators involved in endosomal protein recycling, undergoes truncating mutations that constitute the genetic basis of SYS (2).
Diagnosing SYS involves clinical evaluation and genetic testing, with MAGEL2 gene sequencing conducted if PWS is ruled out (9). The following case report presents a unique clinical scenario in which a patient with SYS experienced recurrent unexplained episodes of high temperatures and seizures, underscoring the neurological features of the disorder. Genetic testing leading to an SYS diagnosis emphasizes the importance of considering this rare genetic condition in patients with hypotonia, atypical febrile episodes, and neurodevelopmental delays. Notably, this is the first reported case of SYS in Iran.
2. Case Presentation
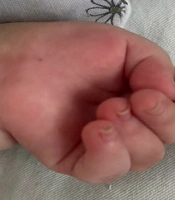
A one-year-old female born at full term to consanguineous parents presented with severe respiratory distress and recurrent episodes of hyperthermia. Breastfed until one year old, she exhibited slower growth than her peers, as measured by the CDC chart, and displayed developmental delay. Severe hyperthermia and respiratory distress had been apparent since birth, manifesting on her first day of life. Physical examination revealed hypotonia, campylodactyly, and hand contractures (Figure 1). A chest X-ray confirmed meconium aspiration, necessitating resuscitation. After treatment for aspiration pneumonia, she was discharged, but episodes of fever and tonic seizures persisted, managed with phenobarbital.
. The image shows symmetrical distal contractures of the hands, evident in the patient's clenched fists with finger flexion deformities, a characteristic feature of campylodactyly. Notable findings also include hypotonia, which contributes to the infant’s limp posture, and small body size relative to gestational age. These findings, alongside respiratory distress, have been present since birth.")
Clinical presentation of an infant with Schaaf-Yang syndrome (SYS). The image shows symmetrical distal contractures of the hands, evident in the patient's clenched fists with finger flexion deformities, a characteristic feature of campylodactyly. Notable findings also include hypotonia, which contributes to the infant’s limp posture, and small body size relative to gestational age. These findings, alongside respiratory distress, have been present since birth.
At one year, the introduction of solid foods coincided with emerging feeding difficulties, leading to recurrent hospitalizations due to respiratory complications, including aspiration pneumonia and chronic hyperthermia. Despite comprehensive treatments and evaluations, her symptoms remained.
The patient was subsequently hospitalized at Mofid Children’s Hospital due to acute respiratory distress, requiring supplemental oxygen and exhibiting increased use of accessory muscles for breathing. During her hospitalization, she received broad-spectrum intravenous antibiotics. Her respiratory status gradually improved over 10 - 14 days, as evidenced by higher oxygen saturation, decreased tachypnea, and resolution on chest x-rays; however, she continued to experience unexplained episodes of hyperthermia and excessive sweating, which resolved spontaneously without antipyretic therapy.
The physical findings, including hand abnormalities, short stature, and chronic hypotonia from birth, raised suspicion of a genetic disorder. Consequently, genetic testing was performed, including whole exome sequencing (WES), which was confirmed by Sanger sequencing. To exclude hormonal imbalances as a possible cause, we assessed GH, TSH, and T4 levels. Although her GH was at the lower end of the range, thyroid function tests were normal (GH = 4.7 [normal range: 5 - 8], TSH = 3.4 [normal range: 0.55 - 5], T4 = 5.3 [normal range: 4 - 12]). Molecular testing confirmed SYS with mutations in the MAGEL2 gene (c.1923dupCp.V643Gfs*70). Additional probable pathogenic variants were identified in the ASPM and KIF7 genes, and these results were validated by Sanger sequencing (Table 1). This diagnosis attributed the hyperthermia episodes to a genetic condition rather than an infectious cause. The parents received counseling about the characteristics of SYS and were advised on genetic counseling for future family planning. Treatment now involves ongoing monitoring for respiratory issues and managing feeding challenges and hypotonia. Currently, the patient remains under follow-up and has shown no further respiratory or feeding difficulties since her initial hospitalization.
| Gene | Exon | Mutation | Chromosome | Syndrome/Condition | Finding Type |
|---|---|---|---|---|---|
| MAGEL2 | 1 | c.1923dupC, p.V643Gfs*70 | chr1523890966, A>AG | Schaaf-Yang syndrome | Primary finding |
| ASPM | 18 | c.7372_7376del, p.R2458Gfs*59 | chr1197071004, CTTTCT>C | Microcephaly, primary, autosomal recessive | Secondary finding |
| KIF7 | 7 | c.1640dupG, p.R549Afs*40 | chr1590190208, G>GC | Al-Gazali-Busmawa syndrome, hydrocephalus syndrome 2, acrocallosal syndrome, Joubert syndrome 12 | Secondary finding |
Genomic DNA was extracted from the peripheral blood. Whole exome sequencing was used to enrich all exons of protein-coding genes as well as other important genomic regions. Next-generation sequencing was performed on an Illumina Sequencer to generate approximately 100 million reads, with the platform examining > 95% of the targeted regions and achieving a sensitivity above 99%. This test allows for the simultaneous detection of point mutations, insertions, and deletions (> 20 bp). Bioinformatics analysis of the sequencing results was conducted using international databases and standard bioinformatics software.
This sample is heterozygous for a pathogenic variant in the MAGEL2 gene. The variants identified, c.1923dupC, p.V643Gfs*70, were reported in ClinVar as pathogenic/likely pathogenic. Mutations in this gene are associated with Schaaf-Yang syndrome. The proband is also a carrier of two likely pathogenic variants in the ASPM and KIF7 genes.
3. Discussion
Our patient initially presented with significant episodes of hyperthermia and respiratory distress, initially suspected to be infection-related, both at birth and at one year of age. Medical professionals at Hamedan Hospital considered infection to be the most probable cause of her hyperthermia and respiratory distress. To investigate further, they conducted a range of diagnostic tests, including chest X-ray (CXR), lumbar puncture (LP), and flow cytometry. Broad-spectrum intravenous antibiotics were initiated to treat the presumed infection after these tests failed to identify a specific cause. Despite treatment, episodes of hyperthermia persisted.
Upon admission to Mofid Children's Hospital, the patient presented with severe respiratory distress and low oxygen saturation, prompting the continuation of broad-spectrum antibiotic therapy.
Given the patient's persistent hypotonia and neurological symptoms, including atypical posture, genetic testing was initiated to explore possible neurogenetic causes. Although respiratory distress improved, her recurrent fevers persisted and did not respond to antipyretics, suggesting a non-infectious etiology. Following the paraclinical findings, the patient's episodic fever and hyperthermia resolved without antipyretic intervention, further indicating a non-infectious, potentially hypothalamic, dysfunction. Genetic testing and blood sequencing confirmed that SYS was likely responsible for these symptoms. The specific mutation identified (c.1923dupCp.V643Gfs*70) and the patient’s distinctive physique and posture led to this targeted genetic evaluation. Additionally, the patient experienced early-life seizures, a less common symptom in SYS, pointing to broader neurodevelopmental involvement in the disorder.
Seizures, while rare in SYS, have been documented and highlight the variability in clinical manifestations based on genetic variations associated with the syndrome (4). One of the primary challenges in diagnosing SYS is the diversity of symptoms, which can lead to misinterpretation, as seen with the patient’s recurring fevers initially thought to indicate infection, thereby delaying the identification of her genetic condition. Typical features of SYS include neonatal hypotonia, feeding difficulties, intellectual disabilities, developmental delays, and joint contractures (5). This complexity is compounded by the existence of related genetic syndromes, such as PWS, which share symptoms like neonatal hypotonia, developmental delay, and feeding challenges (6). However, SYS often involves a higher prevalence of joint contractures, particularly in the interphalangeal joints, compared to PWS (4). Additionally, while hyperphagia and obesity are observed in both disorders (9), differentiating between these syndromes is crucial, as it directly informs treatment strategies, particularly for neurodevelopmental delays.
A challenge with diagnosing SYS is that patients' persistent fevers and episodes of hyperthermia are often misidentified as infections or inflammatory conditions, leading physicians to pursue costly diagnostic tests to identify a presumed source of infection or inflammation. This misdiagnosis can result in the overuse of antibiotics and other unnecessary treatments, which not only consumes resources but also exposes patients to risks associated with invasive procedures, such as LPs and bone marrow aspiration (BMA).
In recent years, reports on SYS patients have described unexplained hyperthermia, temperature instability, feeding difficulties, and respiratory distress. These symptoms may represent the broader clinical spectrum of SYS (4). The neurological symptoms exhibited by our patient, including seizures, emphasize the importance of considering SYS in cases with unexplained neurodevelopmental abnormalities, consistent with other documented instances of SYS.
Similar to other SYS cases reported in the literature, our patient presented with episodic hyperthermia along with other symptoms. However, in our case, the patient also experienced seizures, a less common symptom in SYS patients. Notably, our patient did not present with scoliosis or any spine abnormalities, and due to her young age, she could not be evaluated for ASD. The patient’s age of one year is on the lower end of the spectrum [1 year old], compared to the average age of SYS patients reported in the literature (mean of 8.1 years) (4). These findings underscore the importance of comprehensive neurological evaluation in patients with SYS, as early detection of neurodevelopmental issues can facilitate more tailored interventions.
These observations highlight the importance of including SYS in the differential diagnosis for patients with non-specific symptoms and unexplained fever or hyperthermia (9). A comparison of the genetic findings in our case with those in the literature shows that, in a 2020 study by Ahn et al., three distinct truncating MAGEL2 mutations were identified. Two patients carried the most common mutation, c.1996dupC, previously reported, while two novel mutations, c.2217delC and c.3449_3450delTT, were also documented (10). In our case, the mutation was identified at the c.1923dupCp.V643Gfs*70 locus, suggesting that such variations may explain the differences in disease presentation across patients. Previous studies on genotype-phenotype correlations suggest that the severity of symptoms may depend on the location of the truncating mutation. For instance, patients with the c.1996dupC mutation tend to experience joint contractures, feeding issues, respiratory problems, and more severe intellectual disabilities or developmental delays (4).
Our case highlights the importance of recognizing SYS as a possible diagnosis in young patients with otherwise unexplained neurological and systemic symptoms, which could lead to more accurate diagnoses and targeted care.
Schaaf-Yang syndrome management requires a multidisciplinary approach that addresses the disorder's diverse manifestations. This includes occupational and physical therapy for motor delays, behavioral interventions and educational therapies for intellectual disabilities, and nutritional support for dietary issues. Growth hormone therapy is often used to treat GH deficiencies and to improve growth characteristics (4, 6). From a neurological perspective, effective seizure management and early intervention for developmental delays, along with ongoing monitoring, are crucial to minimize long-term impacts on the patient's quality of life.
Managing recurrent respiratory infections and persistent hyperthermia is particularly challenging in SYS. Frequent fevers can lead to unnecessary antibiotic use and invasive procedures. Hypotonia, aspiration, and recurrent respiratory infections are among the more severe complications, sometimes necessitating early interventions like tracheostomy or intubation, which can further complicate the patient’s condition (4). The rarity and broad spectrum of clinical presentations in SYS pose significant diagnostic and management challenges for healthcare providers (2). Due to the syndrome's uncommon nature, identification can be difficult, especially when physicians lack familiarity with the condition without comprehensive genetic testing (9).
We encourage other clinicians and researchers to expand on our work in identifying various manifestations of this relatively unknown disease. Raising awareness about SYS can help the medical community differentiate it from similar syndromes and disorders. Understanding SYS can aid in recognizing symptoms that may present in future cases, guide the confirmation of SYS through genetic testing (particularly in families with prior cases), and reduce adverse effects in affected individuals. Such awareness also enables more efficient allocation of time and resources by minimizing the misdiagnosis of infections linked to episodic hyperthermia and fosters the development of specific, effective treatments or preventive measures.
Limitations in our study include the inability to fully assess ASD, a common concern in SYS children, due to our patient's young age and associated neurodevelopmental challenges, such as language impairments. Additionally, the novelty of SYS and the lack of long-term follow-up on symptom progression over time are ongoing challenges in understanding and managing this condition.
3.1. Conclusions
Schaaf-Yang syndrome is a rare neurogenetic disorder characterized by a wide range of clinical symptoms, particularly significant neurological deficits. This case highlights the importance of considering SYS in the differential diagnosis for patients presenting with unexplained fever, seizures, and neurodevelopmental delays. By elucidating the phenotypic diversity of SYS, this report emphasizes the need for early genetic testing to identify underlying neurogenetic factors. Prompt recognition and intervention may help mitigate certain neurological deficits associated with SYS, potentially improving patient outcomes.
