




1. Introduction
Hyaline fibromatosis syndrome is a rare autosomal recessive disease (1). It is characterized by abnormal deposition of amorphous hyaline material into the dermis and other tissues (1). It presents with pearly papules or fleshy nodules on the face, neck, ears, scalp, hands, feet, and perianal regions (1, 2). Additional features may include progressive joint contracture resulting in mobility limitation, severe pain with movement, skeletal muscle atrophy, gingival hypertrophy, thickened skin, coarse facial features, osteopenia, and osteoporosis (3, 4). FTT and protein-losing enteropathy are important complications resulting in decreased levels of immunoglobulins and some cellular and humoral deficiency, which may lead to infection susceptibility and affected survival (5). The severity is variable; individuals with the severe form present in infancy and die in early childhood (previously called infantile systemic hyalinosis); other patients that have later onset of the milder form survive into adulthood (previously called juvenile hyaline fibromatosis) (6, 7). Histopathology of skin lesions demonstrates hyaline material as an amorphous eosinophilic substance in the dermis and spindle-shaped fibroblasts spread in it (8). Molecular genetic testing revealed that mutations in the CMG2 gene (capillary morphogenesis gene 2) are responsible for this autosomal recessive condition (9). CMG2 as a transmembrane protein is expressed during capillary formation and binds to collagen IV through a Von Willebrand factor type A (VWA) domain (10).
2. Case Presentation
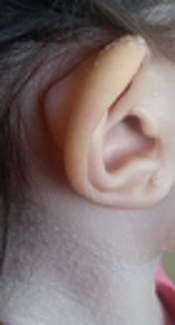
A 2-year-old girl was admitted to Mofid children’s hospital with a history of PID, recently developed whitish skin papules, and subcutaneous nodules around her mouth (Figures 1 and 2). She had been admitted because of upper respiratory infection at the ages of 4, 6, and 9 months and due to severe diarrhea at the ages of 8 and 10 months. Finally, immunologic workup was performed showing the IgG level of 215 mg/dL. The serum level of IgA was 32 mg/dL. The serum levels of IgM and IgE were within the normal ranges. NBT was normal and CD flow cytometry was unremarkable. Then, IVIG was started to control recurrent respiratory infections and poor response to antibiotics. When she was receiving monthly IVIG, her symptoms such as respiratory infections and diarrhea were controlled.

Whitish papules on her neck, face, and ears

Subcutaneous nodules and gingival hypertrophy
She was the only child of healthy consanguineous parents. Her umbilical cord was detached at 25th of life because of omphalitis. At the age of eight months, her parents complained of excessive crying while they were changing her clothes. The shoulder X-ray was unremarkable. Her mother detected multiple slow growing whitish papules appeared on her neck, face, and gum thickening at the age of 12 months. She was receiving regular IVIG since she was 12 months of age. She referred to our department for further investigation to confirm the diagnosis.
We discontinued IVIG and performed immunologic evaluations. The level of serum immunoglobulins was normal (IgM = 35 g/dL, IgA = 58 g/dL, IgG = 680 g/dL). Antibody titers to tetanus toxin and diphtheria were in the normal range. There was no abnormality in peripheral blood flow cytometry. The NBT test was 100% and LTT was normal. Then, CVID diagnosed was ruled out and excisional biopsy of subcutaneous nodule around her mouth was performed. Fibrosis and hyalinization, cells with abundant granular cytoplasm, and spindle-shaped cells spread in the hyaline material suggesting hyaline fibromatosis syndrome were demonstrated. Immunohistochemical (IHC) panel was compatible with fibromatosis (Table 1).
| Results | |
|---|---|
| SMA | Weakly positive in few dispersed cells |
| Desmin | Negative |
| CD34 | Negative |
| CD117 | Negative (cytoplasmic staining in few dispersed cells) |
| Ki-67 | Positive in rare dispersed cell |
| S-100 | Positive |
She underwent four times of surgical excision of hypertrophied gingiva and rectum prolapses. Imatinib was started and could decrease the growth rate of gum and reduce joints contractures.
3. Discussion
CVID is a heterogeneous group of primary immunodeficiency syndromes described as the low level of immunoglobulins and poor or absent specific antibody responses to vaccination (11), in spite of good antibody response in our patient. The symptoms may present at any age but more frequent between 15 and 40 years of age. The low level of IgG (≤ 2 S.D of the mean of the age-adjusted normal range) along with at least low-level IgA or IgM is found in CVID patients (12, 13). Over 90% of adult patients with CVID have IgA levels under the normal range. The level of serum IgM is more variable but it decreases in more than 50% of CVID patients (11). In this patient, a low level of IgG and IgA, recurrent infections, and optimal response to IVIG lead to the diagnosis of CVID. Although antibody response is not reliable at the age of 11 months, it could distinguish THI (transient hypogammaglobinemia of infancy) from CVID. On the other hand, there was no new infection while some new findings like the reduced range of motions and rectal prolapse were revealed in the course of the disease, which is not common in CVID or THI that needs more investigation.
After biopsy and diagnosis of IHF, the decreased level of IgG could be justified by PLE. Of course, it could predispose the patient to recurrent infection. PLE is a more common cause of profound hypogammaglobulinemia (14). The low level of IgA was justified by incomplete synthesis due to the age of the patient. Indeed, the levels of specific antibody responses and isohemagglutinin titers are helpful in the discrimination of THI and PLE from CVID.
The Hyaline fibromatosis syndrome is an AR disease in which the abnormal amorphous hyaline material is deposited in the dermis and other tissues (1). FTT and protein-losing enteropathy are important complications resulting in the decreased levels of immunoglobulins and some cellular and humoral deficiency, which lead to infection susceptibility (5). Other features of IHF including progressive joint contracture resulting in mobility limitation, severe pain with movement, gingival hypertrophy, thickened skin, coarse facial features, whitish skin papules, and subcutaneous nodules were found in this patient.
Indeed, molecular genetic testing to reveal genetic mutations will be confirmatory. When a patient is suspected to PID, the secondary immune deficiency should be ruled out. Moreover, patient follow-up and revaluation until definite diagnosis should be considered.
