


1. Background
Urinary tract infection (UTI) in pregnant women is a common healthcare problem, which is commonly caused by pathogens normally residing in the intestine and genital tract. Occasionally, the diagnosis of UTI is difficult, and many patients are treated based only on signs and symptoms, the error rate was 33%. The microscopy and culture method of diagnosis takes two days for identification and antibiotic sensitivity of bacteria. The patients treated empirically encounter a risk of antimicrobial resistance. The main advantage of molecular diagnosis is identifying the cause within hours up to species level. 16S real-time PCR was narrated by Lehmann et al. using probes specific to a large number of genus/species (1). All bacteria contain the 16S rRNA gene, thereby making the sequencing-based bacterial identification plausible (2). In addition, the 16S rRNA gene comprises variable regions interspersed with nucleotide sequences, which provide a species-specific signature sequence that is the hallmark of bacterial identification. The obtained sequences are compared with the known sequences in the database (3). The method is valuable in the case of a mixture of a wide range of pathogens. Also, it is useful in detecting bacteria that are difficult to grow as well as those in samples obtained from patients’ post-antibiotic treatment. Strain 131 of E. coli has been found to be multi-drug resistant (MDR) (4). Interestingly, MDR ST131 is resistant to fluoroquinolones such as Ciprofloxacin and aminoglycosides like Amikacin and Gentamicin. In hospitalized and community-acquired cases of UTI, strains ST95, 73, 69 of E. coli are frequently isolated and persist in non-extended spectrum beta-lactamase isolates (5). Extra-intestinal pathogenic E. coli, including uropathogenic E. coli, consists of specific phylogenetic groups with different sets of virulence genes and is commonly associated with human diseases.
2. Objectives
This prospective study was carried out in the Department of Microbiology, Patna Medical College, Patna and the Department of Microbiology, GLA University, Mathura. In the following, 16S rRNA gene sequencing was carried out at the BioAxis DNA Research Centre, Hyderabad, India. Urine samples from pregnant women, both asymptomatic and symptomatic, were collected and subjected to screening methods, followed by culture on MacConkey’s media. The antimicrobial sensitivity test was carried out on the nutrient agar.
3. Methods
3.1. DNA Isolation
The isolated bacteria were subjected to DNA isolation using BiopureTM kits (BioAxis DNA Research Centre).
3.2. 16S rRNA Gene PCR
16S rRNA gene was amplified by PCR from the DNA isolated above. The primers used for amplification of the gene were as follows:
• Forward 27F – 5’-AGAGTTTGATCMTGGCTCAG-3’
• Reverse 1492R– 5’-CGGTTACCTTGTTACGACTT-3’
The temperature conditions of PCR were: 5 min at 94°C, 60 sec at 94°C in 35 cycles, 45 sec at 53°C, 90 sec at 68°C and 10 min at 68°C. 4°C was set as hold temperature to keep amplicons safe for the next use.
3.3. Electrophoresis
Electrophoresis of the amplified PCR product was done on 1 kb DNA ladder with 1% agarose gel and TAE as a buffer, which was subsequently visualized by staining with ethidium bromide (Figure 1).

Agarose gel electrophoresis
3.4. Elution
Purification of the PCR product was done by washing with 70% ethanol and sodium acetate, followed by elution from the gel. Electroelution causes rapid and isolation of large fragments of DNA. The DNA band presented in the gel fragment was excised and dialyzed against the TAE buffer. The DNA was precipitated out by an electric current. For separation of agarose from DNA, agarose used is of low melting point agarose is commonly used as it does not denature the DNA structure. Subsequently, the sample was subjected to sequencing.
3.5. Sanger Sequencing Using Dye Terminators
The PCR amplicon was sequenced on ABI 3730XL automated DNA Sequencer. In this method, different fluorescent markers are used for labeling every dideoxynucleotide in a capillary tube. Different colored bands are produced by DNA fragments of different sizes separated in a capillary tube. For a given size DNA fragment, there is a separate band and, the colors indicate different bases at which termination of the fragment has occurred. The bases represented by short fragments, moved first in the capillary. When the light emitted by the laser falls on the capillary tube, the light emitted by the fluorescent dye was recorded on the detector. The signal from the detector of each colored band was analyzed by the sequencer and appeared on the graph as a peak. Each base had a separate peak.
3.6. BLAST Reference
The assembled DNA sequence was used to carry out BLAST with the nr database from NCBI.
3.7. Phylogenetic Tree Construction
Top ten similar sequences of BLAST were retrieved, and a phylogenetic tree was constructed using Clustal omega.
4. Results
The semi quantitative culture technique identified the following bacterial isolates: E. coli, Klebsiella, Pseudomonas, Staphylococcus, coagulase-negative Staphylococcus, and Proteus spp. These were subjected to 16S rRNA gene sequencing, and the results are as follows:
4.1. Sequence Obtained for Sample 1
ATGACCAGCAACACTGGAACTGAGACACGGTCCAGA-CTCCTACGGGAGGCAGCAGTGGGGAATATTGCACAATG-GGCGCAAGCCTGATGCAGCCATGCNGCGTGTATGAAGAA-GGCCTTCGGGTTGTAAAGTACTTTCAGCGGGGAGGAAGG-GAGTAAAGTTAATACCTTTGCTCATTGACGTTACCCGCA-GAAGAAGCACCGGCTAACTCCGTGCCAGCAGCCGCGGTA-ATACGGAGGGTGCAAGCGTTAATCGGAATTACTGGGCGT-AAAGCGCACGCAGGCGGTTTGTTAAGTCAGATGTGAAAT-CCCCGGGCTCAACCTGGGAACTGCATCTGATACTGGCAA-GCTTGAGTCTCGTAGAGGGGGGTAGAATTCCAGGTGTAG-CGGTGAAATGCGTAGAGATCTGGAGGAATACCGGTGGCG-AAGGCGGCCCCCTGGACGAAGACTGACGCTCAGGTGCGA-AAGCGTGGGGAGCAAACAGGATTAGATACCCTGGTAGTC-CACGCCGTAAACGATGTCGACTTGGAGGTTGTGCCCTTG-AGGCGTGGCTTCCGGANNTAACGCGTTAAGTCGACCGCC-TGGGGAGTACGGCCGCAAGGTTAAAACTCAAATGAATTG-ACGGGGGCCGCACAAGCGGTGGAGCATGTGGTTTAATTC-GATGCAACGCGAAGAACCTTACCTGGTCTTGACATCCAC-GGAAGTTTTCAGAGATGAGAATGTGCCTTCGGGAACCGT-GAGACAGGTGCTGCATGGCTGTCGTCAGCTCGTGTTGTG-AAATGTTGGGTTAAGTCCCGCAACGAGCGCAACCCTTAT-CCTTTGTTGCCA
4.1.1. Inference
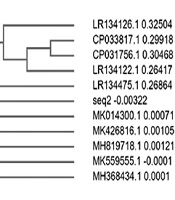
The sequence obtained was 100% identical to the partial gene sequence of 16S rRNA of Escherichia coli strain U5/41 (Figures 2 and 3).

BLAST reference of Escherichia coli

Phylogenetic tree of Escherichia coli
4.2. Sequence for Sample 2
ATGTGCCCAGATGGGATTAGCTAGTAGGTGGGGTAA-TGGCTCACCTAGGCGACGATCCCTAGCTGGTCTGAGAGG-ATGACCAGCCACACTGGAACTGAGACACGGTCCAGACTC-CTACGGGAGGCAGCAGTGGGGAATATTGCACAATGGGCG-CAAGCCTGATGCAGCCATGCCGCGTGTATGAAGAAGGCC-TTCGGGTTGTAAAGTACTTTCAGCGAGGAGGAAGGCGTT-AAGGTTAATAACCTTGGCGATTGACGTTACTCGCAGAAG-AAGCACCGGCTAACTCCGTGCCAGCAGCCGCGGTAATAC-GGAGGGTGCAAGCGTTAATCGGAATTACTGGGCGTAAAG-CGCACGCAGGCGGTCTGTCAAGTCGGATGTGAAATCCCC-GGGCTCAACCTGGGAACTGCATTCGAAACTGGCAGGCTA-GAGTCTTGTAGAGGGGGGTAGAATTCCAGGTGTAGCGGT-GAAATGCGTAGAGATCTGGAGGAATACCGGTGGCGAAGG-CGGCCCCCTGGACAAAGACTGACGCTCAGGTGCGAAAGC-GTGGGGAGCAAACAGGATTAGATACCCTGGTAGTCCACG-CCGTAAACGATGTCGACTTGGAGGTTGTGCCCTTGAGGC-GTGGCTTCCGGAGCTAACGCGTTAAGTCGACCGCCTGGG-GAGTACGGCCGCAAGGTTAAAACTCAAATGAATTGACGG-GGGCCCGCACAAGCGGTGGAGCATGTGGTTTAATTCGAT-GCAACGCGAAGAACCTTACCTACTCTTGACATCCAGAGA-ACTTAGCAGAGATGCTTTGGTGCCTTCGGGAACTCTG
4.2.1. Inference
The sequence obtained was 100% identical to the partial gene sequence of 16S rRNA of Klebsiella aerogenes strain F26 (Figures 4 and 5).

BLAST reference of Klebsiella

Phylogenetic tree of Klebsiella aerogenes
4.3. Sequence for Sample 3
ATGGGCGAAAGCCTGATCCAGCCATGCCGCGTGTGT-GAAGAAGGTCTTCGGATTGTAAAGCACTTTAAGTTGGGA-GGAAGGGCAGTAAGTTAATACCTTGCTGTTTTGACGTTA-CCGACAGAATAAGCACCGGCTAACTCTGTGCCAGCAGCC-GCGGTAATACAGAGGGTGCAAGCGTTAATCGGAATTACT-GGGCGTAAAGCGCGCGTAGGTGGTTCGTTAAGTTGGATG-TGAAAGCCCCGGGCTCAACCTGGGAACTGCATCCAAAAC-TGGCGAGCTAGAGTATGGTAGAGGGTGGTGGAATTTCCT-GTGTAGCGGTGAAATGCGTAGATATAGGAAGGAACACCA-GTGGCGAAGGCGACCACCTGGACTGATACTGACACTGAG-GTGCGAAAGCGTGGGGAGCAAACAGGATTAGATACCCTG-GTAGTCCACGCCGTAAACGATGTCAACTAGCCGTTGGAA-TCCTTGAGATTTTAGTGGCGCAGCTAACGCATTAAGTTG-ACCGCCTGGGGAGTACGGCCGCAAGGTTAAAACTCAAAT-GAATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGGT-TTAATTCGAAGCAACGCGAAGAACCTTACCAGGCCTTGA-CATGCAGAGAACTTTCCAGAGATGGATTGGTGCCTTCGG-GAACTCTGACACAGGTGCTGCATGGCTGTCGTCAGCTCG-TGTCGTGAGATGTTGGGTTAAGTCCCGTAACGAGCGCAA-CCCTTGTCCTTAGTTACCAGCACGTTATGGTGGGCACTC-TAAGGAGACTGCCGGTGACAAACCGGAGGAAGGTGGGGA-TGACGTCAAGTCATCATGGCCCTTACGGCCTGGGCTACA-CACGTGCTACAATGGTC
4.3.1. Inference
The sequence obtained was 100% identical to Pseudomonas entomophila strain 2014 (Figures 6 and 7).

BLAST reference of Pseudomonas

Phylogenetic tree of Pseudomonas entomophila
4.4. Sequence for Sample 4
ATGGGCGAAAGCCTGACGGAGCAACGCCGCGTGAGT-GATGAAGGTCTTCGGATCGTAAAACTCTGTTATTAGGGA-AGAACATATGTGTAAGTAACTGTGCACATCTTGACGGTA-CCTAATCAGAAAGCCACGGCTAACTACGTGCCAGCAGCC-GCGGTAATACGTAGGTGGCAAGCGTTATCCGGAATTATT-GGGCGTAAAGCGCGCGTAGGCGGTTTTTTAAGTCTGATG-TGAAAGCCCACGGCTCAACCGTGGAGGGTCATTGGAAAC-TGGAAAACTTGAGTGCAGAAGAGGAAAGTGGAATTCCAT-GTGTAGCGGTGAAATGCGCAGAGATATGGAGGAACACCA-GTGGCGAAGGCGACTTTCTGGTCTGTAACTGACGCTGAT-GTGCGAAAGCGTGGGGATCAAACAGGATTAGATACCCTG-GTAGTCCACGCCGTAAACGATGAGTGCTAAGTGTTAGGG-GGTTTCCGCCCCTTAGTGCTGCAGCTAACGCATTAAGCA-CTCCGCCTGGGGAGTACGACCGCAAGGTTGAAACTCAAA-GGAATTGACGGGGACCCGCACAAGCGGTGGAGCATGTGG-TTTAATTCGAAGCAACGCGAAGAACCTTACCAAATCTTG-ACATCCTTTGACAACTCTAGAGATAGAGCCTTCCCCTTC-GGGGGACAAAGTGACAGGTGGTGCATGGTTGTCGTCAGC-TCGTGTCGTGAGATGTTGGGTTAAGTCCCGCAACGAGCG-CAACCCTTAAGCTTAGTTGCCATCATTAAGTTGGGCACT-CT
4.4.1. Inference
The sequence obtained was 100% identical to Staphylococcus aureus strain NCTC13616 (Figures 8 and 9).

BLAST reference of Staphylococcus aureus

Phylogenetic tree of Staphylococcus aureus
4.5. Sequence for Sample 5
TTTATGGAGAGTTTGATCCTGGCTCAGGATGAACGC-TGGCGGCGTGCCTAATACATGCAAGTCGAGCGAA
CAGATAAGGAGCTTGCTCCTTTGACGTTAGCGGCGG-ACGGGTGAGTAACACGTGGGTAACCTACCTATAA
GACTGGGATAACTTCGGGAAACCGGAGCTAATACCG-GATAACATTTGGAACCGCATGGTTCTAAAGTGAA
AGATGGTTTTGCTATCACTTATAGATGGACCCGCGC-CGTATTAGCTAGTTGGTAAGGTAACGGCTTACCA
AGGCGACGATACGTAGCCGACCTGAGAGGGTGATCG-GCCACACTGGAACTGAGACACGGTCCAGACTCCT
ACGGGAGGCAGCAGTAGGGAATCTTCCGCAATGGGC-GAAAGCCTGACGGAGCAACGCCGCGTGAGTGATG
AAGGGTTTCGGCTCGTAAAACTCTGTTATTAGGGAA-GAACAAATGTGTAAGTAACTGTGCACGTCTTGAC
GGTACCTAATCAGAAAGCCACGGCTAACTACGTGCC-AGCAGCCGCGGTAATACGTGTGGCAAGCGTTATC
CGGAATTATTGGGCGTAAAGCGCGCGTAGGCGGTTT-CTTAAGTCTGATGTGAAAGCCCACGGCTCAACCG
TGGAGGGTCATTGGAAACTGGGCTTGAGTGCAGAAG-AGGAAAGTGGAATTCCATGTGTAGCGGTGAAATG
CGCAGAGATATTAGTGGAGGAACACCAGTGGCGAAG-GCGACTTTCTGGTCTGTAACTGACGCTGATGTGC
GAAAGCGTGGGGATCAAACAGGATTAGATACCCTGG-TAGTCCACGCCGTAAACGATGAGTGCTAAGTGTT
AGGGGGTTTCCGCCCCTTAGTGCTGCAGCTAACGCA-TTAAGCACTCCGCCTGGGGAGTACGACCGCAAGG
TTGAAACTCAAAGGAATTGACGGGGACCCGCACAAG-CGGTGGAGCATGTGGTTTAATTCGAAGCAACGCG
AAGAACCTTACCAAATCTTGATGAAAACTCTAGAGA-TAGAGCCTTCCCCTTC
4.5.1. Inference
The sequence obtained was 98.47% identical to Staphylococcus saprophyticus strain FDAARGOS_355 (Figures 10 and 11).

BLAST reference of Staphylococcus saprophyticus

Phylogenetic tree of Staphylococcus saprophyticus
4.6. Sequence for Sample 6
TGGGGTTGATCATGGCTCAGATTGAACGCTGGCGGC-AGGCCTAACACATGCAAGTCGAGCGGTAACAGGA
GAAAGCTTGCTTTCTTGCTGACGAGCGGCGGACGGG-TGAGTAATGTATGGGGATCTGCCCGATAGAGGGG
GATAACTACTGGAAACGGTGGCTAATACCGCATAAT-GTCTACGGACCAAAGCAGGGGCTCTTCGGACCTT
GCACTATCGGATGAACCCATATGGGATTAGCTAGTA-GGTGGGGTAAAGGCTCACCTAGGCGACGATCTCT
AGCTGGTCTGAGAGGATGATCAGCCACACTGGGACT-GAGACACGGCCCAGACTCTACGGGAGGCAGCAGT
GGGGAATATTGCACAATGGGCGCAAGCCTGATGCAG-CCATGCCGCGTGTATGAAGAAGGCCTTAGGGTTG
TAAAGTACTTTCAGCGGGGAGGAAGGTGATAAGGTT-AATACCCTTGTCAATTGACGTTACCCGCAGAAGA
AGCACCGGCTAACTCCGTGCCAGCAGCCGCGGTAAT-ACGGAGGGTGCAGGCGTTAATCGGAATTACTGGG
CGTAAAGCGCACGCAGGCGGTCAATTAAGTCAGATG-TGAAAGCCCCGAGCTTAACTTGGGAATTGCATCT
GAAACTGGTTGGCTAGAGTCTTGTAGAGGGGGGTAG-AATTCCATGTGTAGCGGTGAAATGCGTAGAGATG
TGGAGGAATACCGGTGGCGAAGGCGGCCCCCTGGAC-AAAGACTGACGCTCAGGTGCGAAAGCGTGGGGAG
CAAACAGGATTAGATACCCTGGTAGTCCACGCTGTA-AACGATGTCGATTTAGAGGTTGTGGTCTTGAACC
GTGGCTTCTGGAGCTAACGCGTTAAATCGACCGCCT-GGGGAGTACGGCCGCAAGGTTAAAACTCAAATGA
ATTGACGGGGGCCCGCACAAGCGGTGGAGCATGTGG-TTTAATTCGATGCAATGCGAAGAACCTTACCTAC
TCTTGACATCCAGCGAATCCTTTAGAGATAGAGGAG-TGCCTTCGGGAACGCTGAGACAGGTGCTGCATGG
CTGTCGTCAGCTCGTGTTGTGAAATGTTGGGTTAAG-TCCCGCAACGAGCGCAACCCTTATCCTTTGTTGC
CAGCACGTAATGGTGGGAACTCAAAGGAGACTGCCG-GTGATAAACCGGAGGAAGGTGGGGATGACGTCAA
GTCATCATGGCCCTTACGAGTAGGGCTACACACGTG-CTACAATGGCAGATACAAAGAGAAGCGACCTCGC
GAGAGCAAGCGGAACTCATAAAGTCTGTCGTAGTCC-GGATTGGAGTCTGCAACTCGACTCCATGAAGTCG
GAATCGCTAGTAATCGTAGATCAGAATGCTACGGTG-AATACGTTCCCGGGCCTTGTACACACCGCCCGTC
ACACCATGGGAGTGGGTTGCAAAAGAAGTAGGTAGC-TTAACCTTCGGGAGGGCGCTTACCACTTTGTGAT
TCATGACTGGGGTGAAGTCGTAACAAGGTAACC
4.6.1. Inference
The sequence obtained was 100% identical to the partial gene sequence of 16S rRNA of Proteus mirabilis strain NCTC 11938 (Figures 12 and 13).

BLAST reference of Proteus mirabilis

Phylogenetic tree of Proteus mirabilis
5. Discussion
In the present study, a comprehensive molecular characterization of six bacterial isolates was carried out. We observed that the sequence in sample 1 was 100% identical to the partial gene sequence of 16S rRNA of E. coli strain U5/41. Phylogenetically strains included U5/41, E. fergusonii strain ATCC35469, E. fergusonii strain NBRC102419, E. coli strain JCM1649, E. fergusonii strain ATCC35469, Shigella flexneri strain ATCC29903, E. coli strain NBRC102203, Shigella boydii strain P288, Shigella sonnei strain CECT4887, and Brenneriaalni strain pvfi20. Campos et al. also conducted a study in Brazil, of E. coli isolated from urine samples of hospitalized patients and identified strains 131 and 69 as the most frequently found E. coli strains (6). Strain 69 was found to be associated with both community-acquired and healthcare-associated UTIs (7). The MDR manner of these strains was attributed due to the dfrA17-aadA5 gene, which makes these strains resistant to Trimethoprim, aminoglycosides. The other ST groups identified by Campos et al. included ST648, ST405, ST73, and ST10.
The sequence of sample 2 was 100% identical to that of the partial gene sequence of 16S rRNA of Klebsiella aerogenes strain F26. The phylogenetically identified strains were F26, K. aerogenes strain NCTC10006, K. aerogenes strain NCTC10006, K. aerogenes strain FDAARGOS_513 chromosome, K. aerogenes strain CB46l, K. aerogenes strain FDAARGOS_327 chromosome, K. aerogenes strain DAS43, K. aerogenes strain gol2, K. aerogenes strain NCTC9735, and K. aerogenes strain CX-122.
In sample 3, the sequence was 100% identical to that of Pseudomonas entomophila strain 2014. The phylogenetically identified strains were P. entomophila strain 2014, P. entomophila strain 1257 chromosome, Bacterium strain E70 16S ribosomal RNA gene, P. putida strain CK223, P. spp strain AZ5, P. guariconensis strain njensis, P. spp strain BYT-1, P. spp WCHPs060039, P. guariconensis strain MR149, and P. guariconensis strain MR144.
In sample 4, the sequence was 100% identical to that of Staphylococcus aureus strain NCTC13616. Phylogenetically identified strains included S. aureus strain NCTC13616, S. aureus strain NCTC4163, S. aureus strain NCTC11965, S. aureus strain NCTC4137, S. aureus strain NCTC9555, S. aureus strain NCTC5660, S. aureus strain NCTC7121, S. aureus strain NCTC13142, S. aureus strain NCTC12233, S. aureus strain NCTC13552.
In sample 5, the strain identified was Staphylococcus saprophyticus strain FDAARGOS_355. The other phylogenetically identified strains were S. saprophyticus strain FDAARGOS_355, S. saprophyticus strain FDAARGOS_137, S. saprophyticus sub spp. saprophyticus ATCC 15305, S. saprophyticus strain RJ17, S. saprophyticus sub spp. saprophyticus ATCC 15305, Staphylococcus spp. S04009, S. saprophyticus sub spp. saprophyticus strain NCTC7666, S. saprophyticus strain FDAARGOS_336, S. spp 82B, S. saprophyticus strain BVC51.
The sequence of the sample was 100% identical to that of the partial gene sequence of 16S rRNA of Proteus mirabilis strain NCTC 11938. The phylogenetically identified strains were P. mirabilis strain NCTC 11938, P. mirabilis strain PmSC1111, P. mirabilis strain NCTC4199, P. mirabilis strain AR_0029, P. mirabilis strain AR379, P. mirabilis isolate GN2, P. mirabilis strain AR_0156, P. mirabilis strain AR_0159, P. mirabilis strain AR_0059, and P. mirabilis strain AOUC-001.
Jenkins et al. obtained pus samples and joint fluids from 23 patients using 1,343 bp PCR. Of 38 samples using 762/598 bp PCR, 33 samples were negative by both culture and PCR. Moreover, 16S rDNA was identified in 8/17 culture-positive samples (8). The bacteria identified were S. aureus, Streptococcus pneumoniae, Streptococcus viridians, Prevotella pleuritidis, and Prevotella oulorum. Gene sequencing helped in identifying anaerobes in samples positive on culture. As the Gram-positive cell wall is disrupted readily during the extraction process, it is not identified as easily as Gram-negative bacteria (9-12). However, Jenkins et al. found that for both Gram-negative and -positive bacteria, there was no bias as they could not be identified by PCR (8).
Van der Zee et al. demonstrated that PCR-based detection can replace the culture-based diagnosis except in the case of antibiotic sensitivity testing that might be essential for the adequate treatment of patients. These results were confirmed by 16S PCR. However, the limitation of this study was that only a few strains were tested, and hence, the presence and homology of target genes need further substantiation (13).
Tajbakhsh et al. isolated and detected Gram-negative bacteria, causing UTI in patients from Shahrekord Hospitals, Iran. The study used PCR, which was found to be an effective method for diagnosis of bacteria causing UTI, especially Gram-negative ones and also other infections (14).
Abulmeshah carried out a study to identify organisms causing UTI by doing 16S rRNA gene sequencing and BLAST analysis and found that E. coli, K. pneumoniae, S. aureus, P. mirabilis and P. aeruginosa were the most prevalent organisms (15).
5.1. Conclusions
Six bacterial isolates were analyzed by 16S rRNA gene sequencing, followed by the construction of a phylogenetic tree formation up to the species level. This method was a valuable tool for a cost-effective and accurate diagnosis of an array of uropathogens in both asymptomatic and symptomatic pregnant women. If the samples presented MDR on sensitivity testing, the specific strain isolated by PCR would provide guidelines for the management of UTI in pregnant women in the future.
