



1. Introduction
Autosomal recessive congenital ichthyosis (ARCI) belongs to a group of heterogeneous cornification- related disorders with a prevalence of 1/200,000 in the USA (1). The clinical features of ARCI include eclabium, epidermal scaling, alopecia, ectropion, and/or erythema, in addition to collodion at birth (collodion baby). ARCI is divided into harlequin ichthyosis (HI), lamellar ichthyosis (LI), and congenital ichthyosiform erythroderma (CIE) (2, 3). The two latter conditions are at the extreme end of the ARCI spectrum, in which large dark cutaneous scales with negligible erythema are the characteristics of LI while erythroderma and white scales are more frequent in CIE patients (4).
Genetically, ARCI is a heterogeneous disease and results from mutations in 14 genes, including TGM1, ABCA12, NIPAL4, lipo-oxygenase genes (ALOX12B and ALOXE3), LIPN, CYP4F22, ST14, CASP14, SDR9C7, SULT2B1, CERS3, SLC7A4, and PNPLA1 (5-7). However, the genotype-phenotype correlation in ARCI is weak. For example, LI phenotype, frequently but not exclusively, is caused by mutations in the TGM1 gene, but the same mutations can also lead to CIE (3, 6, 7). In this context, several studies have reported that more than 55% of ARCI cases are resulted from germline mutations in the TGM1 gene that encodes transglutaminase-1, a cell membrane-bound epidermal enzyme with a pivotal role in the structure and function of the skin barrier through participating in cell envelope formation (8, 9).
The TGM1 gene has been mapped on the long arm of chromosome 14 (14q12) and encompasses a 14.3 Kb DNA sequence with 15 exons. The gene has a highly conserved transcription start point at exon 2 (8, 10). More than 210 mutations in TGM1 have been identified based on HGMD professional 2019.4 in different ethnic groups. In this context, missense mutations account for most cases, and c.877-2 A > G is the most commonly reported causative mutation in North America (8). Herein, exome sequencing was performed for an Iranian patient, resulting in the detection of a novel homozygous variant of TGM1.
2. Case Presentation
A 9-year-old male patient affected by congenital ichthyosis was referred to our genetics lab, Department of Genetics, Isfahan University of Medical Sciences, in 2020. In clinical examination, the proband showed dry, rough, and scaly skin, hand deformity due to scars, dark parts on knees and elbows, not fully developed ears, notable erythroderma, retruded nasal bridge, nail dystrophy, white scales, loss of eyebrows, and scalp involvement. The proband was a collodion at birth (collodion baby); his parents were first cousins, and the mother had experienced a miscarriage. The two younger siblings were normal with no known history of ichthyosis (Figure 1A).
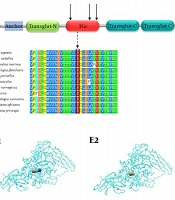
 is homozygous for the c.1165C > T (p.Arg389Cys) variant while the parents and the other sibling are heterozygous for the variant; B, the sequence electropherogram of TGM1: the c.1165C > T (p.Arg389Cys) variant in the proband, father, mother, and sibling (the black box); C, the schematic illustration of the transglutaminase-1 enzyme’s domains and the location of mutations in the protein structure. Anchor, the membrane anchorage region; Transglut-N, transglutaminase family N-terminal domain; TGc, transglutaminase/protease-like homologues domain; Transglut-C, transglutaminase family, C-terminal Ig like domain; D, multiple sequence alignment showed that p.Arg389Cys was located in a highly conserved region. Changes in conserved amino acids have been enclosed in a black box; E, the protein structure modeling of wild-type (E1) and mutated (E2) <i>TGM1</i> (the position of the residue 389 in the protein structure has been indicated). The structures of Arg389 (E3) and Cys389 (E4) residues with their four-angstrom neighbor residues.")
A, the familial pedigree. The proband (II‐2) is homozygous for the c.1165C > T (p.Arg389Cys) variant while the parents and the other sibling are heterozygous for the variant; B, the sequence electropherogram of TGM1: the c.1165C > T (p.Arg389Cys) variant in the proband, father, mother, and sibling (the black box); C, the schematic illustration of the transglutaminase-1 enzyme’s domains and the location of mutations in the protein structure. Anchor, the membrane anchorage region; Transglut-N, transglutaminase family N-terminal domain; TGc, transglutaminase/protease-like homologues domain; Transglut-C, transglutaminase family, C-terminal Ig like domain; D, multiple sequence alignment showed that p.Arg389Cys was located in a highly conserved region. Changes in conserved amino acids have been enclosed in a black box; E, the protein structure modeling of wild-type (E1) and mutated (E2) TGM1 (the position of the residue 389 in the protein structure has been indicated). The structures of Arg389 (E3) and Cys389 (E4) residues with their four-angstrom neighbor residues.
The study was approved by the Review Board of the Isfahan University of Medical Sciences (Grant No. 195122). For sampling, 5 mL of peripheral blood was drawn from the proband and other family members after receiving informed consent. After that, DNA was extracted from blood lymphocytes using a Prime Prep Genomic DNA Extraction kit (GeNet Bio, South Korea), following the instructions of the manufacturer. Then whole-exome sequencing was carried out to detect the possible responsible variant using a next-generation sequencing technology (Novaseq 4000 platform, Illumina, San Diego, CA, USA) with the 100X mean depth of coverage for more than 92% of the target sequence.
Aligning the sequence against the human reference genome (hg19, NCBI Build 38) was performed using Burrows-Wheeler Aligner (BWA) software (https://sourceforge.net/projects/bio-bwa/files/latest/download), and variant calling was performed by GATK software (11, 12). Insertion, deletion, and single nucleotide polymorphisms (SNPs) were sought for using Sequence Alignment/Map tools (SAMtools). Variant filtering was carried out for nonsense, missense, splice site, start codon, stop loss, indel, and frame-shift variants with minor allele frequency (MAF) of < 1% in the 1000 genomes project, exome aggregation consortium (ExAC), NCBI dbSNP version 147, NHLBI GO exome sequencing project (ESP), and Iranome (http://www.iranome.ir/).
We performed Sanger sequencing in a bidirectional manner to confirm the candidate variant derived from WES, followed by co-segregation analysis using exon-specific primers to assess phenotype and genotype segregation in family members. Polymerase chain reaction and Sanger sequencing in the family members were performed using 5’CCTTCTCCCCCTGATACTCC3’ and 5’CAGCTGACAAACCCGTTTAAG3’ primers, and chromatograms were compared against the human reference genome (hg19, NCBI Build 38) via SeqMan software version 5.00 © (DNASTAR, Madison, WI, USA). At the final step, the Human Gene Mutation Database (HGMD) and the literature were investigated to assess the novelty of the candidate variant and investigate its association with ichthyosis.
A homozygous variant, c.1165C > T (p. Arg389Cys), in the exon 8 of the TGM1 gene was observed in the proband (Figure 1B). We also performed Sanger sequencing on the DNA derived from other family members to conduct co-segregation analysis. While one of the siblings was homozygous for the wild-type allele, the other sibling and parents were heterozygous for this variant (Figure 1A).
The variant was referred to as rs757905282 in the NCBI dbSNP database. However, there were no reports on its pathogenicity in the literature nor in the NCBI clinvar database. The mutated allele was found neither in our local exome database (Named GTaC) containing about 1500 exome sequenced samples nor in the Iranome database (www.iranome.ir). As shown in Table 1, in silico analysis was accomplished using mutation taster 2.0 (http://www.mutationtaster.org/) and the FATHMM MKL prediction tool (http://fathmm.biocompute.org.uk/) in conjunction with the evaluation of the variant’s significance at the protein level by SIFT, Polyphen, and PROVEN online software. The variant interpretation was carried out according to the American College of Medical Genetics (ACMG) guidelines. Finally, p. Arg389Cys was predicted as a likely pathogenic variant as it met PM1, PM2, PM5, PP1, PP2, and PP3 ACMG criteria (13).
| Variables | Values |
|---|---|
| Variant | c.1165C>T (p. Arg389Cys) |
| dbSNP rsID | rs757905282 |
| Zygosity | Homozygous |
| ExAC global MAF | 0.000008 |
| PROVEN | Damaging |
| MutationTaster2 | Disease causing (converted rank score: 0.81) |
| FATHMM MKL coding Pred | Damaging (coding score: 0.9366) |
| SIFT | Damaging |
| Polyphen | Probably damaging with a score of 1.000 |
| ACMG variant category | Likely pathogenic |
The modeling of the 3D structure of the transglutaminase-1 protein was performed using phyre2 web-based services for protein structure prediction (http://www.sbg.bio.ic.ac.uk/~phyre2/html/page.cgi?id=index). The PDB files of the wild-type and mutated proteins were generated by phyre2 and UCSF Chimera version 1.13.1 (https://www.cgl.ucsf.edu/chimera/), which were used to construct the 3D structures of both the wild-type and mutant forms of transglutaminase-1 (Figure 1E). On the other hand, since only the incomplete crystal structure of transglutaminase-1 is available at Protein Data Bank (PDB), and the fact that it does not cover the p.Arg389 variant (PDB ID: 2XZZ), we performed protein modeling using the 3D structure of the human factor XIIIa subunit (PDB ID: 1GGT), showing more than 40% similarity in conserved amino acid sequences compared to human transglutaminase-1 (14-16). The Position-Specific Iterative BLAST (PSI-BLAST) of the transglutaminase-1 amino acid sequence against the 1GGT structure of human factor XIIIa showed that most of the conserved residues were distributed in the transglutaminase/protease-like homolog domain at which the p.Arg389 variant was located (16).
3. Discussion
Autosomal recessive congenital ichthyosis belongs to a diverse group of non-syndromic keratinization disorders (7, 17). The most prevalent molecular causes of ARCI are mutations (mainly missense) in the TGM1 gene encoding the transglutaminase-1 enzyme (8). In this study, we investigated the clinical and molecular features of an Iranian ARCI patient. The molecular analysis of the NGS data identified c.1165C > T as a novel missense variant in the TGM1 gene in the proband, leading to the substitution of arginine by cysteine in the residue 389 of the transglutaminase-1 enzyme. This novel variant occurred in a highly conserved position and was predicted to be detrimental based on the conservation scores obtained in various in silico analyses (Table 1).
The transglutaminase-1 enzyme takes part in the development of cornified cell envelope (CCE), a thick physical insoluble barrier of skin cells. The enzyme catalyzes the formation of cross-links between its precursors at the terminal stages of keratinocytes’ differentiation (18). A more thorough understanding of the molecular pathology of ARCI was achieved by observing the absence or deformation of CCE in conditions with reduced activity of the transglutaminase-1 enzyme (8). Therefore, it is believed that transglutaminase-1 is a key player in the normal function of the epidermal barrier, which is weak, porous, and distorted in ARCI patients. This enzyme consists of three transglutaminase domains and one transglutaminase/protease-like homolog domain. The p.Arg389Cys is located in the latter domain, which is a catalytic core domain with 92 amino acids and three critical active sites (Figure 1C).
Missense mutations are responsible for the main pathogenic disease-causing variants of the TGM1 gene, and the rate of benign missense variations is low in this gene. According to the ClinVar database, 140 out of 150 (93.3%) non-VUS (variant of uncertain significance) missense variants of TGM1 are pathogenic, which is higher than the threshold of 51.0% based on ACMG guidelines. Additionally, 197 (55.6%) of the 354 clinical variants of TGM1 reported have been pathogenic, which is also higher than the ACMG threshold of 12.0% (PP2) (13). In addition, this variant was absent in control population databases, including the Iranome and gnomAD genomes (PM2).
The c.1165C > T variant is positioned at a 61 bp hotspot region harboring eight non-VUS coding variants and no benign variants (PM1). On the other hand, according to the PROVEAN software, p.Arg389Cys alteration lowers transglutaminase-1 enzyme activity by generating an unstable protein (8, 19). The substitution of arginine by cysteine can lead to remarkable changes in the conformation of the protein due to the fact that cysteine is a neutral amino acid in terms of electrical charge, so this replacement results in the loss of one positive charge of arginine. In addition, arginine has a bulgy side chain while cysteine is a smaller amino acid. These observations suggest that the arginine at position 389 has a pivotal role in the normal function of the encoded enzyme. In this regard, changes in arginine 389 due to c.1166G > C (p.Arg389Pro) and c.1166G > A (p.Arg389His) mutations have been associated with ARCI in previous reports, suggesting a key role for arginine 389 in the normal function of the transglutaminase-1 enzyme (PM5) (17).
In conclusion, in this study, we described a new variant of the TGM1 gene in an Iranian pedigree, modifying the encoded enzyme’s structure and ultimately leading to congenital ichthyosiform erythroderma. So, our study expanded the number of the known pathogenic variants of this gene, which has molecular diagnostic and genetic counseling implications in Iran.
