1. Introduction
Muscular dystrophy is a degenerative muscle disease in which the muscles that control movement progressively weaken (1). It occurs when one of the genes responsible for the production of proteins involved in myocytes, ion channels, etc., is defective (2). Muscular dystrophies or dysferlinopathies are hereditary disorders that over time engage the patient's various muscles and gradually reduces the ability of the muscle to repair itself by stem cells (3). Limb-girdle muscular dystrophies (LGMDs) are heterogeneous disorders characterized by the weakness of the pelvic and shoulder girdle muscles (4).
Due to genetic heterogeneity and limited data about LGMD genetic makeup in our country, we aimed to investigate the genetic variants in muscular dystrophy using whole-exome sequencing technique following bioinformatics analysis.
2. Case Presentation
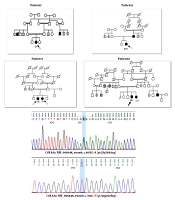
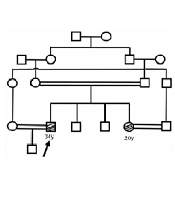
We first identified a family who was referred for genetic counseling for muscular dystrophy. Two members of this family were affected (Figure 1). Their parents had a consanguineous marriage, and these two affected members had weakness of the proximal lower limbs. In the proband, not only weakness of proximal upper limbs but also proximal lower limbs were involved. In the affected individuals, serum creatine kinase (CK) was elevated (reference range: 25 - 200 U/L). The severity of the disease was higher in the patient (proband) at the time of study than the newly-affected sister, such that the proband was wheelchair-bound.

Figure 1.
The pedigree of the referred family. The arrow shows the proband. All the black shapes show the affected individuals.
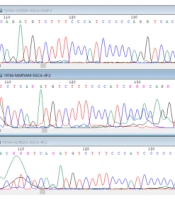
After a physical examination and laboratory testing, due to the probability of a genetic cause and to find the responsible variant, the whole-exome sequencing (WES) technique was requested for the proband. All the participants signed an informed consent. Then, 3 mL of peripheral blood was obtained within the EDTA tubes from all family members. DNA was extracted using standard salting-out protocol. Proband’s DNA sample was sent for exome sequencing (Macrogen, Korea). Data were analyzed, and candidate variants were assessed through online bioinformatics tools and genomic databases such as Varsome (https://varsome.com/) and ClinVar (www.ncbi.nlm.nih.gov/clinvar), respectively. Moreover, variant frequencies were assessed according to the frequencies reported in genomic databases, such as 1000 Genomes Project and ExAC+. For the validation of candidate variants, family co-segregation analyses were performed using Sanger sequencing. Using the online software Primer3 Plus, forward and reverse primers were designed for the candidate variant. All the amplified products were prepared in a final volume of 25 μL for Sanger sequencing (Kawsar Biotech Co., Tehran, Iran). The results were analyzed using CLC workbench 7.7 software.
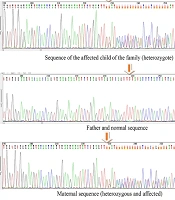
The results of the exome data analysis revealed a variant of c.2864 + 1G > A in the patient, which altered the exon-intron 26 cleavage site in the DYSF gene (Table 1). The assessment of candidate variants in several online prediction tools and databases showed its pathogenicity (Table 1). Genetic changes in this gene are known to be associated with muscular disorders such as LGMD and other dysferlinopathies. The examination of this genetic variant in the patient's sister also showed homozygous mutations. Since the patient's sister was married to a cousin, her husband was also tested for the variant, which was negative (healthy).
Table 1.
Summary of the Candidate Variant Properties
| Gene | Variant | Nucleotide Change | Location | Zygosity | Prediction | Database | Frequency | |
|---|---|---|---|---|---|---|---|---|
| Mutation Taster | ClinVar | Genome AD | ExAC | |||||
| DYSF | rs199954546 | c.2864 + 1G > A | Intron26 | Homozygote | Disease causing | Pathogenic/likely pathogenic | 0.00001 | 0.00001 |
3. Discussion
Here, we reported two LGMD patients and identified the splice site variant. Since the patients’ parents were first cousins, the family showed an autosomal recessive inheritance pattern. Based on the bioinformatics predictions using online tools, this nucleotide change at the splice site may have a deleterious effect on mRNA splicing and the subsequent protein product.
The diagnosis of LGMD may be challenging because no specific biochemical or protein assays are available (5). Dysferlinopathies generally manifest in the late teenage years or primary adulthood. The advancement of dysferlinopathies is usually slow, and most patients lose their ability to walk and run in the second decade or later. The Dysferlin gene placed at chromosome 2p13.1-p13.3, and mutations in this gene cause limb-girdle muscular dystrophy (LGMD) type 2B. Dysferlin protein is commonly expressed in the skeletal muscle, but in affected patients, it is reduced or absent. There are two different types of LGMDs, one is the autosomal dominant, which is called type 1, and the other one is autosomal recessive, that is called type 2. The recessive types are more widespread than dominant ones. The classic form of LGMD type 2B comes with some symptoms like weakness and atrophy in proximal muscles, but some phenotypic variants of this disease and other dysferlinopathies have been described (6). Former cohorts of LGMD done in North China and Northeast China reported that the most common LGMD subtypes were dysferlinopathies (49.52, 38.46%) and LGMD 2A (24.76, 46.15%), followed by sarcoglycanopathies (9.52, 7.69%) and LGMD 1B (6.67, 0%) (7, 8). The frequency of sarcoglycanopathies is different greatly among regions, and the top four LGMD subtypes in Korea did not include sarcoglycanopathies (9, 10).
Apparently, the lack of dysferlin might affect the immunity system. Other authors recommend that splice site mutations in dysferlin might cause a phenotype that is related to muscle inflammation (11).
As exome sequencing clinical usage becomes more widespread, the time to diagnosis is decreasing significantly, resulting in improved treatment strategies and management of genetic disorders. A study reported in 2020 demonstrated that AMP-activated protein kinase (AMPK) γ1 plays a key role in fixing the plasma membrane, and AMPK is essential for possible treatments (12). Another recent study on Korean populations demonstrated that the most common pathogenic variants are nonsense mutations for dysferlinopathies, and about 50% of the studied patients had one nonsense variant. The Korean study concluded that a small molecule drug called Ataluren, which promotes the readthrough of immature termination codon, may be helpful for dysferlinopathies and other muscle-related disorders caused by nonsense mutation (13).
In sum, we described two Iranian LGMD2B patients with recessive mutations in the DYSF gene that is highly disease-causing according to the ACMG criteria. NGS-based techniques such as whole-exome sequencing can identify the molecular genetic basis of the disease in families with limb-girdle muscular dystrophy. The results can be helpful in identifying potential carriers in the family and in prenatal diagnosis to the families involved. Exome sequencing that has recently been merged into the medical genetic diagnostic area has already identified the role of more than thousands of genes, especially in Mendelian disorder.