




1. Background
Galactosemia is one of the metabolic disorders discovered in newborn screening (1). In other words, galactosemia is an inherited disorder of galactose metabolism due to deficiency in one of the three enzymes involved in the Leloir pathway (2). Galactose-1-phosphate uridyltransferase (GALT), galactokinase, and uridine diphosphate glucose (UDP)-galactose-4-epimerase are members of the Leloir pathway of galactose metabolism (3). The deficiency of any of the three enzymes causes galactose accumulation in plasma which causes galactosemia.
Classic galactosemia (CG) is caused by mutations in the GALT gene. In CG, The GALT enzyme, which is responsible for conversion of galactose-1-phosphate (Gal-1-P) and UDP-glucose into glucose-1-phosphate and UDP-galactose, loses its activity.
The GALT deficiency leads to the accumulation of Gal-1-P in various organs leading to the clinical presentations. The CG is an autosomal recessive disturbance of galactose metabolism (4, 5). The treatment of CG is diet limitations of lactose-containing foods (6-8). The CG occurs with a frequency of approximately 1.2 in 10,000 to 1 in 60,000 live births (9). The incidence rate of CG in southern of Iran is 1 in 28,000 live births (10). The GALT gene is located on chromosome 9p13 and contains 11 exons (11). The CG is heterogeneous at clinical and molecular levels; accordingly, 336 different mutations have been identified in recent years (12, 13).
Several common mutations are detected in the GALT gene. The most prevalent observed is the Q188R mutation, which has been reported to account for 54 - 70% of CG (13-15). The Q188R, L195P, and K285N mutations are common (16, 17) in Caucasians and whites; however, the S135L mutation in African populations has been reported as the common mutation (18-20). The N314D variant is a common change in Duarte galactosemia (21). The identification of mutations involved in the development of CG in theeach population can play an essential role in early diagnosis and intervention (22, 23).
2. Objectives
This study aimed to identify the profile of GALT gene mutations in Iranian patients with galactosemia. Furthermore, this study provides some data for the investigation of the introduction of new mutations of the GALT gene that involved the cause of galactosemia in the Iranian population.
3. Methods
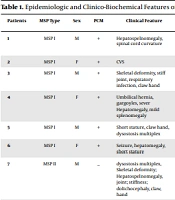
This cross-sectional study was carried out on the newborns of Tehran province, Iran, whose samples were referred to the newborn laboratories of clinics and hospitals under the supervision of Tehran University of Medical Sciences, Iran, from March 2014 to December 2019. In this study, 31 patients diagnosed with galactosemia were investigated for the mutations of the GALT gene. The demographic and clinical data, such as age at diagnosis, gender, diarrhea, liver failure, hepatomegaly, jaundice, vomiting, cataracts, hemolytic anemia, sepsis status, and GALT activity (μmol/h per gHb), were collected using a questionnaire by asking the children’s parents (Table 1). The GALT enzyme activity in red blood cells was assessed by the Beutler test. The normal range was defined as 37 - 66 μmol/h per gHb.
Table 1.Demographic and Clinical Data of Patients with Galactosemia
| PN | Gender | Age at Diagnosis (m) | Diarrhea | Liver Failure | Hepatomegaly | Jaundice | Vomiting | Cataracts | Hemolytic Anemia | Sepsis | GALT Activity (IU)* |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 1 | + | + | + | + | + | + | - | - | 13.0 |
| 2 | F | 2 | + | - | + | + | - | - | - | - | 12.5 |
| 3 | M | NS | - | - | - | + | - | - | - | - | 9.5 |
| 4 | F | 2 | + | + | + | + | + | - | - | - | 29.0 |
| 5 | F | NS | + | + | + | + | + | + | + | + | 16.0 |
| 6 | M | 3 | + | + | + | + | - | - | - | - | Undetectable |
| 7 | F | 2 | - | - | - | - | - | - | - | - | 13.2 |
| 8 | F | 3 | + | + | + | + | - | - | - | - | 17.1 |
| 9 | M | 2 | + | - | - | - | - | + | - | - | 7.0 |
| 10 | F | NS | + | + | + | + | + | - | + | - | 19.5 |
| 11 | M | NS | + | - | - | - | - | - | - | - | 6.2 |
| 12 | F | 5 | + | + | + | + | + | - | - | - | Undetectable |
| 13 | F | 3 | + | + | + | + | - | - | + | - | Undetectable |
| 14 | F | 3 | + | - | - | - | - | - | - | - | 1.0 |
| 15 | M | 4 | - | - | - | - | - | - | - | - | 12.0 |
| 16 | M | NS | + | + | + | + | + | + | - | + | 9.4 |
| 17 | F | 4 | + | + | + | + | - | - | - | - | 4.7 |
| 18 | M | NS | + | + | - | + | - | - | - | - | 6.3 |
| 19 | F | 2 | - | + | - | - | - | - | - | - | 14.0 |
| 20 | M | 3 | + | + | + | + | - | - | - | + | 11.0 |
| 21 | M | 2 | + | - | - | - | - | - | - | - | Undetectable |
| 22 | F | NS | + | - | - | - | - | - | - | - | 1.7 |
| 23 | F | 2 | + | + | + | + | + | + | + | + | Undetectable |
| 24 | M | 3 | + | - | - | - | - | - | - | - | Undetectable |
| 25 | F | 2 | + | - | - | - | - | - | + | - | 23.3 |
| 26 | M | 2 | + | + | + | + | + | + | - | - | 7.0 |
| 27 | F | 3 | + | + | + | + | - | - | - | + | 9.7 |
| 28 | F | 2 | - | - | - | - | - | - | - | - | 14.0 |
| 29 | M | 3 | + | + | + | + | + | - | - | - | 2.3 |
| 30 | F | 4 | + | + | + | + | - | + | + | + | Undetectable |
| 31 | M | NS | + | + | + | + | - | - | - | - | 5.1 |
Abbreviations: PN, patient number; F, Female; M, Male; NS, newborn screening; m, months; y, years; IU, international unit for enzyme’s catalytic activity.
For the investigation of the variants of the GALT gene in patients with galactosemia, all patients were subjected to exons sequencing of the GALT gene; accordingly, the blood samples were collected from each patient in tube-containing ethylenediaminetetraacetic acid. Genomic deoxyribonucleic acid (DNA) was extracted from peripheral blood white cells via a genomic DNA extraction kit (Add Bio Inc., Korea) following the manufacturer’s procedure. Eight pairs of primers were designed for sequencing the GALT gene, covering all 11 exons of the gene (Table 2). The polymerase chain reaction (PCR) products were sequenced with the Sanger sequencing method in an automatic sequencing system ABI 7500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The results were investigated using FinchTV software (version: 1.4.0; Geospiza, Seattle, WA, USA). The results of sequencing were compared to the reference sequence in the GenBank database. Finally, the type and frequency of GALT gene mutations were evaluated in Iranian patients with galactosemia.
Table 2.Primer Sequence
| Primer Number | Exon Number | Sequence 5' - 3' | PCR Product |
|---|---|---|---|
| 1 | E1 and E2 | F: 5'- AAAGTGAAAGGTGAGGCACG -3' | 750 bp |
| R: 5'- TGACCCAGAAGGAGGTTCAC -3' | |||
| 2 | E3 and E4 | F: 5'- GCCTGTCCAGTCTTTGAAGC -3' | 350 bp |
| R: 5'- GGTTTGAAAGTTGTAAGAGGGG -3' | |||
| 3 | E5 | F: 5'- CACAGCCAAGCCCTACCTCTC -3' | 190 bp |
| R: 5'- ACCTCACAAACCTGCACCCAA -3' | |||
| 4 | E6 | F: 5'- CTTTTGGCTAACAGAGCTCCG -3' | 200 bp |
| R: 5'- TTCCCATGTCCACAGTGCTGG -3' | |||
| 5 | E7 and E8 | F: 5'- ACCTGCCTGTTCTTCTCTGC -3' | 700 bp |
| R: 5'- TACTGGGAGCAACCTCCATC -3' | |||
| 6 | E9 | F: 5'- GCTGAGAGTCAGGCTCTGATTCC -3' | 155 bp |
| R: 5'- CCAGAAATGGTGTTGGGGCT -3' | |||
| 7 | E10 | F: 5'- GGGTTTGGGAGTAGGTGCT -3' | 320 bp |
| R: 5'- GGGCAACAGAAGTATCAGGT -3' | |||
| 8 | E11 | F: 5'- GAAGTCCATGCCACCATTCT -3' | 230 bp |
| R: 5'- TTCAAGGCCCTTTCTGCTTA -3' |
Abbreviations: PCR, polymerase chain reaction.
4. Results
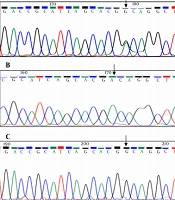
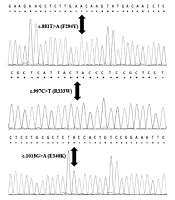
The average age of diagnosis in this study was 2 months, and 58% of patients were female. However, 42% of patients were male. The PCR sequencing analysis identified 11 different pathogenic mutations on the GALT gene in Iranian patients, including five mutations in exon 10, two mutations in exon 9, two mutations in exon 5, one mutation in exon 6, and one mutation in exon 7. Nevertheless, no mutations were detected in exons 1, 2, 3, 4, and 8. All detected mutations were homozygous and missense types. According to Figure 1, this study identified six new mutations of the GALT gene in the Iranian population, namely c.442C>T (R148W), c.881T>A (F294Y), c.997C>T (R333W), c.940A>G (N314D), c.1030C>A (Q344K), and c.1018G>A (E340K). Other mutations (Figure 2) identified in this study were c.855G>T (K285N), c.563A>G (Q188R), c.404C>T (S135L), c.626A>C (Y209S), and c.958G>A (A320T). The most common mutations in the current study were p.Q188R (51.6%), K285N (9.67%), E340K (9.67%), and R148W (6.45%). Table 3 shows a summary of GALT gene mutations. The patients who carried mutations in the GALT gene showed reduced GALT enzyme activity, compared to the normal range (Tables 1 and 3).

Figure 1.
Electropherograms of new GALT mutations identified in Iranian population
Table 3.GALT Mutations in Iranian Patients with Galactosemia
| PN | Nucleotide Change | Mutation Type | Amino Acid Change | Region | GALT Genotype | Frequency (%) | Status of Mutation Report in Iranian Population |
|---|---|---|---|---|---|---|---|
| 1 and 2 | c.442C>T | Missense | R148W | 5 | R148W/ R148W | 6.45 | NM |
| 3 | c.881T>A | Missense | F294Y | 9 | F294Y/ F294Y | 3.22 | NM |
| 4, 5, and 6 | c.855G>T | Missense | K285N | 9 | K285N/ K285N | 9.67 | PR (26) |
| 7 | c.997C>T | Missense | R333W | 10 | R333W/ R333W | 3.22 | NM |
| 8 | c.940A>G | Missense | N314D | 10 | N314D/ N314D | 3.22 | NM |
| 9 | c.1030C>A | Missense | Q344K | 10 | Q344K/ Q344K | 3.22 | NM |
| 10,11, and 12 | c.1018G>A | Missense | E340K | 10 | E340K/ E340K | 9.67 | NM |
| 13-28 | c.563A >G | Missense | Q188R | 6 | Q188R/ Q188R | 51.6 | PR (26) |
| 29 | c.404C>T | Missense | S135L | 5 | S135L/ S135L | 3.22 | PR (26) |
| 30 | c.626 A>C | Missense | Y209S | 7 | Y209S/ Y209S | 3.22 | PR (26) |
| 31 | c.958 G>A | Missense | A320T | 10 | A320T/ A320T | 3.22 | PR (26) |
Abbreviations: PN, patient number; NM, new mutation; PR, previously.

Figure 2.
Electropherograms of other GALT mutations identified in Iranian population
5. Discussion
Galactosemia is one of the metabolic disorders resulting from the deficiency of GALT activity (1, 3). This study identified 11 different pathogenic mutations of the GALT gene in Iranian patients. The R148W, F294Y, R333W, N314D, Q344K, and E340K mutations of the GALT gene were identified as new mutations in the Iranian population; no report of these mutations has been presented in previous studies. The other five K285N, Q188R, S135L, Y209S, and A320T mutations identified in this study were previously reported by Mirzajani et al. (24, 25) in the Iranian population.
The results of the current study showed that Q188R mutation had the highest frequency in the population of Iranian patients (51.6%). After the Q188R mutation, the most common mutations observed in this study were K285N and E340K. The aforementioned results are similar to the results Mirzajani et al. reported in 2006 among Iranian patients (25). The aforementioned results are also similar to the results of other studies. Seyrantepe et al. studied the genetic variations of GALT in patients with galactosemia in the Turkish population, and the results also showed that the most common mutations were Q188R and K285N (26).
In another study, Zekanowski et al. evaluated mutations in the GALT gene in 40 patients with galactosemia in Poland. The results showed that the two Q188R (51%) and K285N (33%) mutations were the most common mutations detected in the aforementioned population (27). Crespo et al. examined galactosemia-related mutations in the GALT gene in the patient population in Argentina and reported that the most common mutations were Q188R and K285N (28). In addition, a comprehensive study published by Tyfield et al. in 1999 showed that the most common GALT gene mutation in patients with galactosemia is Q188R, which is the most common mutation in numerous populations (17). Q188R is generally associated with a severe biochemical phenotype. In vitro expression analysis has confirmed that the mutation results in a substantial, if not complete, loss in GALT activity.
Another mutation observed in this study was c.442C>T, which converts the amino acid arginine to tryptophan. This mutation was never reported in Iranian patients and was classified as a new mutation in this study. In similar studies, Reichardt et al. reported this mutation in the United States, which results in the formation of an unstable polypeptide and an inefficient enzyme (29). The identification of R148W, F294Y, R333W, N314D, and Q344K mutations in Iranian patients was interesting since it has been previously reported that this mutation is very common among European and South American populations (11, 26, 28, 30). S135L mutation was identified in the current study and the study conducted by Mirzajani et al. (25) in the Iranian population. This mutation is also encountered in patients of African origin (4, 19, 20). Lai et al. reported that S135L mutation in the GALT gene is a prevalent cause of galactosemia among black patients, and GALT activity varies in different tissues of patients homozygous for S135L (18).
A320T and Y209S mutations were also detected with similar frequency (3.22%), which are likely to be family-specific. The aforementioned mutations were previously reported in the Iranian and European populations (25, 31). In the current study, the patients who carried mutations in the GALT gene showed reduced GALT enzyme activity, compared to the normal range; this finding is consistent with the results of a study conducted by Garcia et al. (6) related to GALT enzyme activity. Routine screening tests for galactosemia help diagnose the disease quickly, manage symptoms after a positive test, carry out a careful clinical assessment quickly, and perform genotyping to determine whether the patient will be kept on a galactose restriction diet.
One of the limitations of this study is the small number of the studied samples, which suggests that this test should be performed on a larger population. In addition, as previously mentioned in the background, there are other genes in the path of galactose metabolism that cause different types of galactosemia; therefore, it is recommended to check the sequence of these genes in patients.
5.1. Conclusion
Classic Galactosemia is an inherited metabolic disease with life-threatening symptoms in newborn. Nevertheless, early diagnosis can be managed the symptoms and mortality. The disease caused by a severe deficiency of the enzyme galactose-1-phosphate uridylyltransferase. Mutations in this gene can cause a defect in this enzyme. Identification of these mutations can play an important role in early diagnosis and disease management. It is necessary to perform galactosemia screening despite difficulties and complexities. This study identified 11 different pathogenic mutations on the GALT gene in the Iranian population with galactosemia. The identification of the mutations involved in the development of CG in the Iranian population can play an essential role in early diagnosis and intervention. The GALT gene mutations identified in this study can be used as screening markers to identify Iranian children with CG.