

1. Background
Phenylalanine hydroxylase (PAH; EC1.14.16.1), a hepatic enzyme that requires tetrahydrobiopterin (BH4) as a catalytic coenzyme, is responsible for converting L-phenylalanine to L-tyrosine (1). Phenylalanine hydroxylase (PAH) deficiency results in elevated phenylalanine levels in blood, body fluids, and the nervous system (1). One of the most important clinical symptoms of PAH deficiency is mental retardation (2). Other prevalent symptoms are involuntary and rhythmic movements, seizures, microcephaly, and skin disorders such as eczema, skin pigmentation, and musty body odor (1). Some genetic defects impair the liver's ability to produce phenylalanine hydroxylase, resulting in an accumulation of phenylalanine in tissues such as the brain and exerting several damaging effects (2). The other impact of phenylalanine hydroxylase enzyme absence is lack of tyrosine synthesis, resulting in defective melanin pigment formation and ensuing phenotypes such as blond hair, blue eyes, and lack of hair pigment (3). Investigations of the three-dimensional structure of the PAH enzyme indicate that it contains four monomers. Each monomer has three structural domains, including a regulatory domain at the amino terminus (amino acids 1-142), a catalytic domain (amino acids 143-410), and a tetramerization domain at the carboxyl end (amino acids 452-411) (4). The gene responsible for encoding the PAH enzyme is located on the long arm of chromosome 12 at q12-q24.1 and consists of a 90 kb region on this chromosome (5). Phenylalanine hydroxylase (PAH) gene contains 12 introns and 13 exons, encoding 2.5 kb mRNAs (6). The most extended exons and introns are exon 13 and intron 2, with 892 and 17874 bp, respectively (7). Hitherto, more than 520 mutations have been identified in the PAH gene (6). In addition to the mutations, the PAH gene has many polymorphisms used in carrier identification and prenatal diagnosis (8). These polymorphisms are divided into RFLPs, multiple allelic markers, and single nucleotide polymorphisms (SNPs) (9). One of the frequent outcomes of PAH gene mutation is a disease name phenylketonuria. Phenylketonuria (PKU; OMIM 261600) is an inborn metabolic defect that results in reduced phenylalanine amino acid metabolism in the body (10), which occurs due to the mutation of the phenylalanine hydroxylase enzyme gene (1). All types of PKU are considered autosomal recessive diseases (11). The incidence of phenylketonuria in white populations is 1/10000 per live birth (12). The prevalence of PKU in Iran is 1/3627 per live birth, which has been reported to be the outcome of a high percentage of consanguine marriages (13). However, the statistics information varies from population to population (12, 14, 15).
2. Objectives
Despite a bunch of research concerning PAH mutations, much need still exists to recognize mutations in the aforementioned gene in different societies and establish practical treatment strategies. Therefore, this study aimed to determine the PAH mutations in Khuzestan province families with PKU.
3. Methods
3.1. Subjects
The study subjects comprised patients with phenylketonuria who attended the Noor Medical Laboratory of Ahvaz. Two families were included in this study; the first and second families consisted of 3 and 4 members, respectively.
Figures 1 and 2 represent the genealogy chart of two families. Blood samples were obtained with consent from subjects into an EDTA tube and stored at -70°C until test performance. The conditions for entering the clinical diagnosis test by a specialist doctor, medical tests, and checking the level of phenylalanine and the patient's consent, and in the absence of the above, the sample was not used in the research.

Family booklet no. 1

Family booklet no. 2
3.2. DNA Extraction
The modified phenol-chloroform method was used to extract DNA from white blood cells. 1000 µL cell lysis buffer was used to lysis the 500 µL blood sample. The mixture was centrifuged at 4500 rpm for 4 minutes or at 7000 rpm for 2 min, and the precipitate was mixed with 500 μL of cell lysis buffer until it was completely dissolved. The final volume was brought up to 1500 µL. After adding 300 µL nuclei lysis buffer, the mixture was incubated at room temperature for at least 10 minutes until no clumps were seen. In the next step, 100 µL saturated NaCl (6 molar) and 600 µL chloroform were added, and the mixture was shaken and centrifuged at 4500 rpm for 4 minutes or at 7000 rpm for 2 minutes. 300 µL of the supernatant was separated and poured into a 1.5 µL microtube. Then, 1000 μL of cold 96% ethanol was added to the supernatant, and the mixture was slowly shaken until the DNA coil started to appear. The mixture was centrifuged at 1000 rpm for 2 minutes to precipitate the DNA coat. Incubation at room temperature or 56°F to dry was followed by adding 50 l TE or diH2O and incubating for 15 to 20 minutes until the precipitate was completely dissolved.
3.3. Polymerase Chain Reaction
Polymerase chain reaction (PCR) method was used to investigate the mutation spectra of exons 1 to 11 and exon-intron regions in the PAH gene of 7 members of two families with children whose test for PKU was positive. Amplifications were carried out at a final volume of 20 µL, including 200 ng DNA sample, 10 µL Taq DNA Polymerase 2x Red Master Mix, which contained 0.4 mM of each dNTP and 1.5 mM MgCl, (Ampliqon, ID No.5200100, Denmark), 0.1 pM of each primer, and 6.0 µL dH2O. The nucleotide sequence of primers is shown in Table 1. The amplification stage was as follows: the initial cycle was at 95°C for 5 min followed by 35 subsequent cycles of 30 s at 95°C, 30 s at 60°C and 30 s at 72°C and the final extension at 72°C for 5 min. Since there are prevalent mutations in some exons, we classified them into three groups group A (exons 3, 6, 7, 11, and 12), group B (exons 1, 2, 8, 9, and 10), and group C (exons 4, 5, a13, b13 and c13), with the prioritization trend from groups A to C to perform PCR method and detect PKU-related mutations. Table 1 represents the specific primer sequences.
| Forward | Reverse | |
|---|---|---|
| PAH 1AF, PAH 1AR | 5'-CAGCAAGGCAGTGTCTTAG-3' | 5'-CAGAGAGTTTCCTGCCCAAG-3' |
| 1B | 5'-TCCTCCTAGTCGAGGTTA-3' | 5'-AACCCCCTAAAGCAATGGTC-3' |
| 2 | 5'-TGATCATTTAATTGCCCTGGA-3' | 5'-GCCTGTTCCAGATCCTGTGT-3' |
| 3 | 5'-AACTAACTGCCCCACCTCCT-3' | 5'-AATCCCCCAAACAGTCTTCC-3' |
| PAH4 | 5'-CCGGTTTCTAAGGAAAATGGA-3' | 5'-TAATTTTTCCCAGCCCTCGT-3' |
| 5 | 5'-AGCAAGGAGGCTCATGCTAA-3' | 5'-GCACATGAACACATGCACAC-3' |
| 6 | 5'-CCGAAAGTGAGAGCAAATCC-3' | 5'-TTCCTGGAGGAATCAACCTG-3' |
| 7 | 5'-TGCCTAGCGTCAAAGCCTAT-3' | 5'-TGGATCACACAGGATGACCAAA-3' |
| 8 | 5'-CCTACCCTGCACCTGTCTCT-3' | 5'-ATGCTTGATCTCCGAAATGG-3' |
| 9 | 5'-GAAAACAAAGGGCCAAGTGA-3' | 5'-TGCAAATGTAACCCACCACA-3' |
| 10EX | 5'-CCACTGACTCACATGCCAAT-3' | 5'-ACCCACAGCCATCATCAAAT-3' |
| 11 | 5'-GGGTAGAACCTAGTATGTGC-3' | 5'-GGGAGAGAAACTGTCTATGG-3' |
| PAH 12F, 12R | 5'-CTCTAGGGAGGTGTCCGTGT-3' | 5'-GGAAGGTCAAAGGAAAAAGG-3' |
| 13A | 5'-TGCTTTGCACTGAGGACACT-3' | 5'-TGGGCTTCTGACTTAAAGAAAA-3' |
| 13B | 5'-AAAATTTGGGACCTGCTTCA-3' | 5'-CCTGCAAAGGGTTGGAGATA-3' |
| 13C | 5'-TTTTCTTTAAGTCAGAAGCCCATT-3' | 5'-CCTGCAAAGGGTTGGAGATA-3' |
Specific Primer Sequences
3.4. Electrophoresis
In order to analyze the PCR product, we used agarose gel electrophoresis. In order to conduct this procedure, 3 µL of the PCR product was electrophoresed on agarose gels (1.5 %) and then photographed, using a safe stain to detect a single band at an appropriate concentration. To sequence the product of PCR, the remainder was delivered to the sequencing section.
4. Results
In the current study, exons 1 to 11 and exon-intron regions of the PAH gene in a three-member family (family number 1) and a four-member family (family number 2) were evaluated. The result of extracted DNA quality has shown in Figure 3, representing a specific 288 bp band of the PCR products on 1.5% agarose gel.

PCR product electrophoresis, group B exons on 1.5% agarose gel
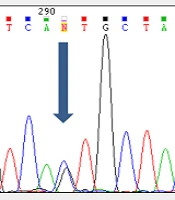
Analyzing the nucleotide sequences of exons 1 to 11 and exon-intron regions in the PAH gene of the study population led to the identification of two IVS10-11 G > A and G > C IVS2 + 5 mutations in introns 10 and 2, respectively. Interestingly, identified G>C IVS2 + 5 (Accession number LC611730.1) mutation in family number 1 and IVS10-11 G>A mutation in family number 2 were found to be heterozygous in parents and homozygous in the children with PKU. Parents in both studied families had also found a kin relationship (cousin-cousin kinship). Moreover, characteristics of family number 1 and 2 with the two identified mutations are set out in Tables 2 and 3, respectively. Figures 4 and 5 also represent the electropherograms for the two determined mutations, respectively.
| Patient | Parents | Phenotype | Mutation in Both Alleles G>C IVS2 + 5 |
|---|---|---|---|
| Father | Relative | Healthy | - / + |
| Mother | Non-relative | Healthy | - / + |
| Child | Relative | cPKU a | + / + |
Characteristics of Patients with G>C IVS2+5 Mutation in Family No. 1
| Patient | Parents | Phenotype | Mutations in Both Alleles G>A IVS10-11 |
|---|---|---|---|
| Father | A distant relative | Healthy | - / + |
| Mother | Non-relative | Healthy | - / + |
| Child | Relative | cPKU a | + / + |
| Child | Relative | Healthy | - / - |
Characteristics of Patients with G>A IVS10-11 Mutation in Family No. 2
 mutation in family number 1, which corresponds to (A) of the father of the family; (B) of the mother of the family; and (C) of the child with the PKU in the respected family (sequence sequenced).")
Electropherogram of G>C IVS2 + 5 (Accession number LC611730.1) mutation in family number 1, which corresponds to (A) of the father of the family; (B) of the mother of the family; and (C) of the child with the PKU in the respected family (sequence sequenced).
 of the father of the family; (B) of the mother of the family; (C) of the child with PKU; and (D) of the child in another family (gone sequence).")
Electrophoregram for IVS10-11 G>A mutation in the family number 2, which corresponds to (A) of the father of the family; (B) of the mother of the family; (C) of the child with PKU; and (D) of the child in another family (gone sequence).
5. Discussion
This study aimed to assess the mutation spectra of exons 1 to 11 and exon-intron regions in the PAH gene of 7 people from families of two children with PKU. The current study's findings indicate two G>C IVS2 + 5 and IVS10-11 G>A mutation sequences in the family number 1 and 2, respectively. The presence of these two mutations in a homozygous form in children has led to the occurrence of the classical PKU disease phenotype.
The current findings of our study are consistent with those of Bonyadi et al., who examined 13 different PAH mutations in 44 people with PKU in Azeri-speaking northwestern areas of Iran and reported a high frequency of IVS10-11G>A mutation (19.3%) in the respective population (16).
Undoubtedly, genetic disorders are capable of causing severe financial and emotional problems in societies. Phenylketonuria, an autosomal recessive disorder, is the most widespread amino acid metabolism defect with various biochemical and clinical manifestations (17-19). Except for children with PKU who have been treated with a low phenylalanine diet, this disease can cause severe mental retardation in patients who have not been cured (17). Therefore, there are some limitations in terms of treatment, which increase the necessity of a neonatal screening program that identifies couples at risk of having a baby with PKU (17). Therefore, to prescribe prompt treatment, the prevalence and spectrum of PAH gene mutations should be monitored in patients with PKU (17).
The (c.1066-11) IVS10-11 G>A mutation occurs by replacing G with A at position 546 of intron 10 in the PAH gene, located in the 11 bp upstream region of the intron ten and exon 11 boundaries. This mutation activates a secreted splicing site and causes the in-frame insertion of a 9-nucleotide template between exons 10 and 11 in the processed mRNA. If such mRNA is translated, an inactive protein would be formed with three additional amino acids (Gly-Leu-Gln) (20).
Moreover, our findings further support the idea of Haerian Ardakani et al., who examined 26 PKU patients in Golestan province and showed that the IVS10-11G> A mutation has a high prevalence (19.23%) in the studied population (21). Another study which is in agreement with our results examined the exons 10, 7, 6, and 11 of the PAH gene in 31 patients with phenylketonuria from Khorasan Razavi province and indicated that IVS10-11G> A mutation, as the most common mutation in the study subjects, has 19% frequency (22).
The IVS10-11G>A mutation is the most common PKU disease in Mediterranean countries and has been reported as a relatively highly prevalent mutation in countries such as Iran 24.6% (12), Greece 12.5% (19), Turkey 32% (23), Egypt 30.8% (24) and Saudi Arabia 17.5% (21). This mutation is also called the Mediterranean mutation (21). Given the high frequency of this mutation in Mediterranean countries, it can be inferred that this mutation might have originated in eastern Mediterranean countries and then spread to other regions through population migration or genetic drift (21). In light of the high frequency of IVS10-11G>A mutation in different societies, it may be considered an identification marker in genetic screening for at-risk individuals.
One unanticipated finding of the present study was the observed G>C IVS2 + 5 (c.168 + 5 G>C) mutation with the accession number of LC611730.1, which was located in intron 2 of the PAH gene of family number 1. Interestingly, this mutation has not been reported previously, and therefore, it can be considered a new mutation in the PAH gene. Various studies have reported new mutations in the PAH gene of different societies, of which we can mention Zschocke (15) and Lee et al. (25) research works.
Zschocke identified 29 mutations in the PAH gene in European populations. These mutations were found to be R408W in haplotype 2 in Eastern Europe, IVS10-11G> A in the Mediterranean countries, IVS12 + 1G> A in Denmark and England, Y414C in Scandinavia, I65T in Western Europe, and R480W in haplotype 1 in the British Isles (15).
Lee et al. have also investigated the PAH gene in 79 Korean patients. They identified 39 different mutations, of which ten mutations were new. Of the new mutations, 9 were missense mutations (P69S, G103S, N207D, T278S, P281A, L293M, G332V, S391I, A447P) and R243Q, IVS4-1G> A, and E6-96A> G mutations were found to be the most common ones comprising 32% of all mutations (25).
5.1. Conclusions
Two common (IVS10-11G> A) and novel (G>C IVS2 + 5) identified mutations could be used to achieve a better understanding of PKU disease and to figure out how to cope with the disease symptoms. With the aid of the findings of this study, it can be possible to recognize the most effective treatment strategy for children with PKU.
