Epidermolysis bullosa includes a broad and inhomogeneous spectrum of congenital genetic and blistering diseases. These diseases are different in terms of severity, prognosis, clinical and histological appearance, and inheritance pattern, but they all have the common feature of blistering due to trauma and its exacerbation in hot weather. These disorders can be divided into three general headings and subcategories: Epidermolysis bullosa simplex (EBS), junctional, and dystrophic (
1).
This disease is divided into three main groups according to the location of the blister involving the skin membrane, which are:
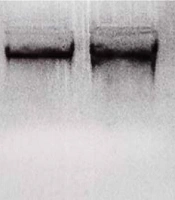
- Simplex Epidermolysis bullosa (EBS): It is the most common type of EB and has an autosomal dominant inheritance pattern. It usually heals without scarring, and only in the epidermis area is the skin cracked and blistered, mainly affecting the palms and feet.
- Junctional Epidermolysis bullosa (JEB): This disorder is characterized by the formation of blisters in the area of the central lamina lucid or basement membrane (where the dermis joins the epidermis). Its inheritance is autosomal recessive, and disruption of the connections between the epidermis and the basement membrane leads to blisters in the upper part of the basement membrane.
- Dystrophic Epidermolysis bullosa (DEB): Signs and symptoms of DEB vary from person to person. In mildly infected cases, the blisters mainly involve the knees, elbows, hands, and feet. In more severe cases, extensive blisters can cause vision loss, disfigurement, and other health problems (
2).
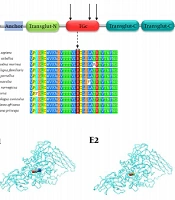
In the DEB type, the C7 collagen protein does not function properly, and the COL7A1 gene, which encodes the C7 protein, is mutated. This protein is the main component of the fibers attached to the epidermis and helps strengthen the skin's middle layer. By disrupting this type of protein or reducing it, the layers of the skin are not properly connected to each other (
3). The National Epidermolysis Bullosa Registry estimated that the overall prevalence of Epidermolysis bullosa in the United States is 11.1 per one million live births, with an incidence of one in every 51,000 live births (
4). The incidence of this disease is the same in different races, and it is seen equally in both genders (
5).
The inheritance of the dystrophic type of this disease is dominant and recessive. In childhood, erosions of the esophagus and its narrowing may cause scarring of the oral mucosa and permanent flexion contraction of the joints secondary to their scarring, and the adhesion of the fingers may significantly disrupt their quality of life (
6).
Corresponding cell carcinoma and infection are the two leading causes of patients being grounded and dying. Although the sensitivity of the skin to trauma decreases with age in these patients, its permanent and progressive changes make their life prognosis unfavorable (
7). Cloning of the human collagen type 7 gene, COL7A1, revealed that this gene consists of 118 exons, and the total size of the human COL7A1 gene is 32 kilobases (31132 bp from the start point to the polyadenylation site) (
8).
This gene encodes an mRNA of about 8.9 kilobases, and its location has been mapped on the short arm of human allele 3, region 21.1. 3p. Genetic studies show a mutation in the COL7A1 gene responsible for creating type VII. In DEB (dominant and recessive type), it has been proven that type VII collagen is responsible for the coordination of collagen fibrils in the tissue, and the presence of a mutation in the above gene causes collagen fibrils to disintegrate (
9).
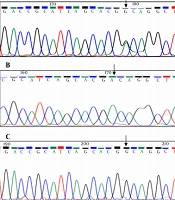
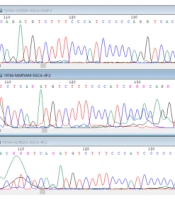
In fact, more than 600 distinct mutations have been identified in the COL7A1 gene, which includes a range of mutations, such as mutations that cause a premature termination codon, resulting from nonsense mutations, small deletions or additions, splicing junction mutations, with the ability to shift the translational reading frame. It includes other meaning mutations that are more difficult to identify. In fact, the general relationship between genotype and phenotype has been confirmed (
10).
According to the above information and the presence of a genotype-phenotype relationship, the types of mutations in the COL7A1 gene in five families with EB in Khuzestan were investigated to compare the mutations in the COL7A1 gene in these five families with EB in Khuzestan with the reported cases.