

1. Background
Epidermolysis bullosa includes a broad and inhomogeneous spectrum of congenital genetic and blistering diseases. These diseases are different in terms of severity, prognosis, clinical and histological appearance, and inheritance pattern, but they all have the common feature of blistering due to trauma and its exacerbation in hot weather. These disorders can be divided into three general headings and subcategories: Epidermolysis bullosa simplex (EBS), junctional, and dystrophic (1).
This disease is divided into three main groups according to the location of the blister involving the skin membrane, which are:
- Simplex Epidermolysis bullosa (EBS): It is the most common type of EB and has an autosomal dominant inheritance pattern. It usually heals without scarring, and only in the epidermis area is the skin cracked and blistered, mainly affecting the palms and feet.
- Junctional Epidermolysis bullosa (JEB): This disorder is characterized by the formation of blisters in the area of the central lamina lucid or basement membrane (where the dermis joins the epidermis). Its inheritance is autosomal recessive, and disruption of the connections between the epidermis and the basement membrane leads to blisters in the upper part of the basement membrane.
- Dystrophic Epidermolysis bullosa (DEB): Signs and symptoms of DEB vary from person to person. In mildly infected cases, the blisters mainly involve the knees, elbows, hands, and feet. In more severe cases, extensive blisters can cause vision loss, disfigurement, and other health problems (2).
In the DEB type, the C7 collagen protein does not function properly, and the COL7A1 gene, which encodes the C7 protein, is mutated. This protein is the main component of the fibers attached to the epidermis and helps strengthen the skin's middle layer. By disrupting this type of protein or reducing it, the layers of the skin are not properly connected to each other (3). The National Epidermolysis Bullosa Registry estimated that the overall prevalence of Epidermolysis bullosa in the United States is 11.1 per one million live births, with an incidence of one in every 51,000 live births (4). The incidence of this disease is the same in different races, and it is seen equally in both genders (5).
The inheritance of the dystrophic type of this disease is dominant and recessive. In childhood, erosions of the esophagus and its narrowing may cause scarring of the oral mucosa and permanent flexion contraction of the joints secondary to their scarring, and the adhesion of the fingers may significantly disrupt their quality of life (6).
Corresponding cell carcinoma and infection are the two leading causes of patients being grounded and dying. Although the sensitivity of the skin to trauma decreases with age in these patients, its permanent and progressive changes make their life prognosis unfavorable (7). Cloning of the human collagen type 7 gene, COL7A1, revealed that this gene consists of 118 exons, and the total size of the human COL7A1 gene is 32 kilobases (31132 bp from the start point to the polyadenylation site) (8).
This gene encodes an mRNA of about 8.9 kilobases, and its location has been mapped on the short arm of human allele 3, region 21.1. 3p. Genetic studies show a mutation in the COL7A1 gene responsible for creating type VII. In DEB (dominant and recessive type), it has been proven that type VII collagen is responsible for the coordination of collagen fibrils in the tissue, and the presence of a mutation in the above gene causes collagen fibrils to disintegrate (9).
In fact, more than 600 distinct mutations have been identified in the COL7A1 gene, which includes a range of mutations, such as mutations that cause a premature termination codon, resulting from nonsense mutations, small deletions or additions, splicing junction mutations, with the ability to shift the translational reading frame. It includes other meaning mutations that are more difficult to identify. In fact, the general relationship between genotype and phenotype has been confirmed (10).
According to the above information and the presence of a genotype-phenotype relationship, the types of mutations in the COL7A1 gene in five families with EB in Khuzestan were investigated to compare the mutations in the COL7A1 gene in these five families with EB in Khuzestan with the reported cases.
2. Methods
2.1. Research Subjects
The patients studied in this research were five families with EB who had been referred to Norgen Ahvaz Medical Genetics Laboratory in 2018-2019. The inclusion criteria for entering the study included symptoms of EB disease, confirmation of the disease phenotype by a specialist doctor, and the patient's informed consent to participate in the study. The entrance of people who did not have skin symptoms common to this disease and were assigned by a different specialist doctor to EB disease was limited.
2.2. Sample Collection and Preparation
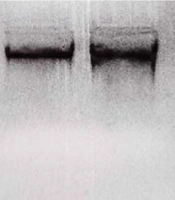
After collecting clinical information and obtaining consent from the families, genetic counseling and genealogy of individuals were done. First, 5 mL of peripheral blood was taken from the patients and poured into the Falcon containing 0.5% EDTA anticoagulant. The saturated salt method was used to extract DNA. After DNA extraction, the quality of DNAs was calculated by quantitative and qualitative methods. A Nanodrop device was used for quantitative analysis, and electrophoresis was used for DNA qualitative analysis (Figure 1).

Photo gel of DNA extraction products shows bands generated from human DNA. The first band created is the 500 bp band, where low molecular weight DNA fragments are deposited. The next bands of DNA fragments are heavier. The bands on the left are indicators or ladders.
2.3. PCR
The sequence of primers used in this study is shown in Table 1. To perform PCR, 10 microliters of Red Mix solution were added to 0.5 microliters of R and F primers. Then, 7.5 microliters of distilled water and 2 microliters of purified DNA were added in the last step. After PCR, its products were electrophoresed (Figure 2). In the next step, the products were placed in the ABI 3130XL sequencer for sequence reading and finally analyzed using the Chromas software at Blast NCBI and ENSEMBL sites.
| Exon and Primer | Sequence | Melting Temperature | GC% |
|---|---|---|---|
| Ex-35-36 | |||
| Forward | 5’-CTC ACC CGC TAT TTG GAT TT-3’ (20 mer) | 56.4 | 45 |
| Revers | 5’-ATG GGG GAG TCA CAG AT-3’ (20 mer) | 58.4 | 50 |
| Ex-37-38 | |||
| Forward | 5’-CCT GAT GAG TGC CAT GTG AT-3’ (20 | 58.4 | 50.0 |
| Revers | 5’AGA ACT ATG AAG CCC AGC AC-3’ (20 mer) | 58.4 | 50.0 |

The gel photo of PCR products shows the successful PCR of exons 36 - 38 of the COL7A1 gene and the deposition of each of the exons of this gene in different bands that are separated and precipitated based on molecular weight.
3. Results
This study examined five families from different parts of Khuzestan province. After genetic counseling and drawing their family tree, the types of mutations in these families were evaluated. Drawing the genealogy of all five families showed repeated family marriages over several generations. The pedigree of one of the five families is shown as an example in Figure 3.

Drawing the genealogy of all five families shows repeated family marriages over several generations. The pedigree of one of the five families is shown as an example.
The results showed that the common mutation p.P1411fs /T c.4233del was observed in four families, resulting from the deletion of thymine nucleotide (T) (Figure 4).

The results of Chromas software in codon 1411 show the deletion of thymine nucleotide in exon 37 of the COL7A1 gene by changing the sequence of proline amino acid.
Also, in this study, a new mutation that has not been reported so far was identified, which is c.6269del C / p.P2090fs del C, and the deletion of the C nucleotide led to the change of PPGPK amino acid sequence to PLAP (one family) (Figure 5). This family includes a parent with a relationship (cousin-cousin) and two children, a boy and a girl with EB, both of whom died.

The results of Chromas software analysis at codon position 2090 in exon 36 of COL7A1 gene; cytosine nucleotide has been deleted. The result is the change of PPGPK amino acids to PLAP.
Examining the chromatogram of this studied family led to the identification of a proband carrying a heterozygous change. The results of chromatogram analysis related to this proband using Chromas software showed the deletion of cytosine nucleotide. This deletion causes a dominant change.
Examining the chromatograms of patients from four other studied families led to identifying the proband with a similar heterozygous change. The chroma chromatogram analysis revealed an amino acid change caused by single nucleotide deletion. As seen in Figure 3, thymine nucleotide deletion followed by the change of proline amino acid was observed.
4. Discussion
Epidermolysis bullosa (EB) is a group of rare genetic diseases that cause fragile skin in sufferers. This disease is caused by mutations in the gene coding for structural proteins located in the basement membrane (BMZ) area of the skin. The lack of these proteins or their failure to function leads to loose connections between the dermis and the epidermis, leading to loss of skin integrity (11).
In Iran, the incidence rate of EB is not precisely known, and due to the unfavorable economic and social conditions in areas such as Khuzestan province, it is not well diagnosed. However, the prevalence of EB types with autosomal recessive inheritance is expected to be high, with a rate of about 38% of consanguineous marriages. This increases the possibility of passing it on to future generations. However, much effort is being made today to diagnose this disease, interpret the molecular results, and combine them with its diagnosis processes (12). At least 15 genes associated with EB are known to cause different forms of this disease. Numerous mutations in these genes have been identified, and the severity of clinical symptoms of the disease depends on the type of mutation, the pattern of inheritance, and the location of the mutation in the gene (13).
Epidermolysis bullosa, especially the dystrophic type, is inherited with both autosomal dominant and recessive inheritance patterns (14). Consanguinity is common in the genetic background of patients with very rare conditions, as seen in the study population with EB in the Iranian population and the families with consanguineous relationships. Therefore, limiting or preventing consanguineous marriages seems useful, even if neither parent has symptoms of a particular disease (15).
In 2010, Galehdari et al. identified a new homozygous single nucleotide deletion in codon 2090 of the COL7A1 gene in exon 74 (c.6269-6270delc). They used the direct sequencing method of 118 exons of the COL7A1 gene to prove the diagnosis of DEB. They were a 10-year-old Iranian girl. Based on the clinical symptoms and family history, this girl was suspected of having DEB. The patient's parents were related to each other in the third degree and were heterozygous for the same mutation and, therefore, mandatory carriers. There was no family history of similar symptoms in the proband. This family did not exist. This mutation causes a premature termination codon in codon 2116 located in collagen subunits. The lack of skin biopsy made it impossible to differentiate DEB forms. This study is the first on Iranian patients with DEB. It was done and confirmed using molecular diagnosis (16).
In 2012, Wertheim-Tysarowska et al. identified 26 different mutations, 13 of which were new, using direct sequencing of 118 exons of the COL7A1 gene (17). According to the research conducted on exons 36-38, we found a new and most likely pathogenic c.6269 del C mutation in exon 36 as a heterozygous mutation that has not been reported so far.
4.1. Conclusions
Considering that the COL7A1 gene has been observed in Khuzestan province, it is expected that the knowledge of these new mutations will help diagnose prenatal screening carriers. Also, it is essential to use new kits with new primers based on known mutations to detect new ones. Screening and genetic counseling before marriage in families where affected people have been seen should be considered necessary.
