




1. Introduction
Limb-girdle muscular dystrophy (LGMD) is genetically heterogeneous. Its clinical symptoms include weakness of the hip and shoulder muscles in the earlier phases, followed by involvement of the proximal part muscles of the arms and legs (1). Studies investigating the relationship between genes and the disease in large families and families with consanguineous marriages have shown that at least 11 different loci are responsible for the limb-girdle muscular dystrophy and that the mutations in genes encode sarcoglycan proteins are associated with this disease (2).
Estimating the prevalence of limb-girdle muscular dystrophy is difficult because the disease has multiple features overlapping with those of other muscle diseases. The prevalence ranges from 1 out of 14,500 individuals to 1 out of 123,000 individuals (3).
Mutations in different genes cause several types of limb-girdle muscular dystrophy. These genes encode proteins involved in muscle repair and protection. Some of the proteins encoded by these genes cluster with other proteins to form larger protein complexes (4, 5).
Limb-girdle muscular dystrophy can have different inheritance patterns. The most common types of this disease are inherited in an autosomal recessive trait. Other types of LGMD have an autosomal dominant pattern of inheritance (6, 7).
This study aimed to report a case of muscular dystrophy with a disease-causing mutation in the limb girdle gene (SGCA). To this end, whole-exome sequencing (WES) was performed to identify the likely disease-causing genetic mutations in a consanguineous Iranian family affected by LGMD. This study also aimed to present the first LGMD case caused by an SGCA deletion mutation, which was not described previously.
2. Case Presentation
In this study, an Iranian family with a 10-year-old boy suffering from congenital muscular dystrophy was examined. The inclusion criterion for the study was the confirmation of the disease symptoms provided by a specialist doctor, and the exclusion criterion was the absence of the clinical features related to the disease. The patient had a history of hereditary muscular dystrophy. He was the first child from a consanguineous marriage (Figure 1). The patient was referred to a medical geneticist due to muscular dystrophy disorder (Figure 2). Electromyography (EMG) performed on the right upper and lower extremities showed a myopathic pattern. A muscle biopsy of the right quadriceps showed variable-size muscle fibers, fiber necrosis, and fiber regeneration. Large hyaline hypereosinophilic fibers were scattered inside the vesicles. There was crowding of nuclei in the center, with multinucleation, degeneration of the fibers, and fatty infiltration consistent with muscular dystrophy.

Pedigree of the studied family. Genealogy of the patient referred to the laboratory due to limb-girdle disease
. Muscular analysis of the patient's leg area. Thinning of the thighs and pseudo-hypertrophy (Left).")
Patient 1 in the presented study was a 10-years old boy: Muscular atrophy of the patient’s neck area and also atrophy of both shoulders with the loss of both deltoids and muscular analysis of the patient’s left shoulder blade (Right). Muscular analysis of the patient's leg area. Thinning of the thighs and pseudo-hypertrophy (Left).
2.1. Whole-Exome Sequencing
After extracting DNA from the buffy coat based on the FAVORGEN manufacturer’s protocol (Biotech Corp, Cat. No.: FABGK 001, Taiwan), the concentration and quality of the DNA samples were analyzed using a Thermo NanoDrop One (Thermo Fisher, USA) at an absorption of 100 - 200 ng/μL and a ratio of 1.8 - 2.0 at 260/280 nm. WES was only performed on the proband. According to the results of the data analysis, there was a novel deletion mutation (p.A107fs:c.319-329del) located in exon 10 (NM_000023.4) of the SGCA gene, but there were no mutations in other genes. As for MutationTaster and SIFT at p.A107fs, the following was proposed:
The c.319-329del variant was pathogenic. Additionally, the Exome Aggregation Consortium (ExAC) and 1000 Genomes databases were used to identify the rare variants (frequency < 1%) seen, specifically in the afflicted juveniles. The reported SGCA gene mutations were compiled based on the Human Gene Mutation Database.
2.2. Polymerase Chain Reaction
The polymerase chain reaction (PCR) was performed using a Bio-Rad thermal cycler as follows: 12.5 μL Master Mix 2X (Thermo Scientific), 1 μL DNA, 0.5 μL forward primer, 0.5 μL reverse primer, and H2O to a final volume of 25 μL. Genomic DNA was PCR-amplified with the forward primer 5’-CCT AGG AGC CCT ATG AAT-3’(20 mer) and the reverse primer 5’-CAT TCC CTG AGA GCC TG TTG-3’(21 mer). The PCR reaction was performed as follows: initial denaturation at 95°C for 5 minutes, followed by 35 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 60 seconds (8, 9).
2.3. Sanger Sequencing
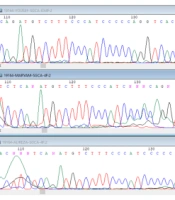
In the present study, a case of autosomal recessive LGMD with a homozygous nonsense mutation (p.A107fs:c.319-329del) in the SGCA gene was detected in a 10-year-old boy in an Iranian family descended from heterozygous and carrier parents (Figure 3). Furthermore, it was shown that this method may have facilitated detecting the rare causative genetic variants in patients with limb-girdle (10).
 (C: afflicted son). The sequence of the child of codon 304, which shows the deletion of phenylalanine in exon 10, which is in the red circle, is characteristic of heterozygous form, and the time of displacement of an adenine instead of guanine in codon 349.")
Sequencing data analysis. The patient carries a homozygous nonsense mutation (p.A107fs:c.319-329del) (C: afflicted son). The sequence of the child of codon 304, which shows the deletion of phenylalanine in exon 10, which is in the red circle, is characteristic of heterozygous form, and the time of displacement of an adenine instead of guanine in codon 349.
3. Discussion
In this study, the WES method was adopted to detect the causative gene defects associated with LGMD in an Iranian family. The index patient was a homozygous carrier for p.A107fs mutation (in SGCA), and his parents were both heterozygous. Therefore, the NM_000023.4 (SGCA): c.319-329del mutation was confirmed to be responsible for LGMD-phenotype in the proband. This was the first Iranian case of an LGMD phenotype caused by SGCA gene mutation. In the study by Balci et al., proximal muscle (midbody) analysis was determined as the most common etiologic factor involved in the disease (33%) (11). The incidence of limb-girdle muscular dystrophy in Western countries ranges from 1 in 500,000 to 1 in 100,000. However, studies in the Indian subcontinent have shown that the prevalence of this disease is much higher in Southeast Asia (approximately 1 in 30,000 - 50,000). Data from eight medical centers in India have also indicated that classical galactosemia is one of the most important causes of neonatal cholestasis (12). According to Ben Hamida et al., however, this condition was the second most common cause in the given regard (23%) (13). In a study by Biancheri et al., all children (0 - 12 years old) with clinical features of limb-girdle muscular dystrophy were examined (14). The patients were followed up for six months after controlling the disease and discharging them. The most common symptoms were muscle atrophy of the neck muscles in 17 subjects and muscle atrophy of the leg muscles in 12 subjects, and 7 out of 17 subjects did not respond to the treatment (14). In a study by Carss et al., the most common disease of limb-girdle muscular dystrophy was recorded in the neonatal period with cholestasis (15).
Finally, this detected deletion SGCA mutation may have been a rare mutation hotspot carried by the patients’ ancestors. Taking into account our study results and the patient’s family history, it was suggested that this genetic mutation should be included in genetic testing platforms for LGMD cases.
3.1. Conclusions
In the present study, a case of LGMD with a homozygous deletion mutation (p.A107fs:c.319-329del) in the SGCA gene was reported. This substitution was found to cause a premature termination of the phenylalanine protein by converting the adenine instead of guanine in codon 349 to a stop codon, which may have created a major problem in the phenylalanine protein. Moreover, it was revealed that the WES method may have facilitated the detecting of rare causative genetic variants in patients with LGMD.
