

1. Background
Genetic disorders of the skeletal system encompass a wide range of conditions, with over 450 known cases identified. Regarding this group of diseases, because it is heterogeneous and more than 300 genes are involved in its development, achieving a definite diagnosis in the field of skeletal genetic disorders and determining the genes related to it is of great importance (1).
Charcot-Marie-Tooth (CMT) disease stands out as one of the most prevalent inherited neuropathies, affecting a significant portion of the population with a varied prevalence of 1 in 2500 to 1 in 9200 live births, dependent on factors such as ethnicity, region, and gender (2). This disorder is part of a wide-ranging, diverse, and intricate cluster of genetically determined neuropathies, showcasing patterns of autosomal dominant, autosomal recessive, or X-linked inheritance. Though at least 51 genes have been previously associated with this condition, the actual anticipated number falls within the range of 50 to 100 genes (3). Clinical symptoms of CMT typically encompass muscle weakness, atrophy, loss of sensation, and reduced musculoskeletal reflexes. Additionally, CMT is often categorized into 2 main types: CMT1 (demyelinating) and CMT2 (axonal), distinguished based on upper limb motor conduction velocities (MCV) (4). The molecular diagnosis of CMT poses significant challenges due to several key factors: (1) the diverse manifestations resulting from a single mutation, (2) the complex genetic spectrum of etiology, and (3) the considerable overlap in symptoms with amyotrophic diseases (5, 6). These complexities highlight the intricate nature of CMT and emphasize the need for sophisticated diagnostic approaches to address its multifaceted genetic and clinical presentation.
These factors indeed exemplify the complex and diverse nature of CMT disease. Therefore, it is crucial to comprehend the molecular causes of CMT for patients, disease diagnosis, and genetic counseling. Meanwhile, the identification of genetic triggers for CMT is invaluable, as many new treatments target specific genes. Notably, among CMT genetic disorders, PMP22 duplication/deletion, along with pathogenic variants of MFN2, GJB1, and MPZ, rank among the most common causes (7). As DNA sequencing technologies advance, particularly with the widespread use of whole-exome sequencing (WES), our genetic knowledge of CMT is expanding. Molecular diagnosis can now be made in 60% of CMT cases using WES (8-10). This technique has become a cornerstone in identifying mutated genes, as pathogenic mutations predominantly occur in the coding regions of genes (exons), which represent a mere 1% of our entire genome. While WES has proven instrumental, it is important to acknowledge the advantages of whole-genome sequencing (WGS) over other diagnostic techniques. Whole-genome sequencing offers unparalleled coverage of coding, non-coding, internuclear, and mitochondrial genome regions. Additionally, it enables the identification of structural variants and facilitates dense homozygosity mapping at the genome level (5). Through exome testing, we can pinpoint the causes of skeletal genetic disorders and, through genetic examination of carriers, prevent the transmission of pathogenic mutations to the next generation. Various studies demonstrate that by developing an exome test panel encompassing all relevant genes associated with these skeletal diseases, an accurate, rapid, and cost-effective molecular diagnosis can be achieved. This is a significant stride forward in our quest to dissect and understand the genetic intricacies of sensory-neural disorders in the shortest possible time and with the highest achievable accuracy.
2. Methods
The present cross-sectional study examined a family referred to the Noorgen Medical Genetics Laboratory in Ahvaz due to neuro-motor disorders in 2022 - 2023. The nucleus of the family includes the father, mother, a healthy female child, and a 20-year-old male child displaying the CMT phenotype, with observed symptoms of muscle weakness in the arms and legs, as well as curvature of the spine. Genealogical research revealed that, on the maternal side, the maternal aunt had been suffering from thanatophoric dysplasia and had passed away at the age of 50 due to a heart attack. Inclusion criteria were the confirmation of clinical manifestations and imaging results by a specialist doctor and the patient's consent to participate in the experiment. Initially, peripheral blood samples (5 mL) were collected from the patients in EDTA tubes. DNA extraction was performed using the salting out method, followed by quantitative (NanoDrop) and qualitative evaluation (agarose gel) of the extracted DNA. The Illumina HiSeq 2500 platform was used to determine the genome sequence. Once the sequencing data was obtained using the WES method, the next step involved analyzing the results. Trench sequencing was then used to confirm the variant. The sequencing of primers used in the research is shown in Table 1.
| Primer | Sequence |
|---|---|
| LRSAM1-EX25-F-376 | GTG TGG CAA GGA GAG CAC TT |
| LRSAM1-EX25-R-376 | CAC GTT CAG AGA CAA CCC TGT |
Sequence of Primers Used for the Polymerase Chain Reaction of the LRSAM1 Gene
The polymerase chain reaction (PCR) was carried out in a final volume of 20 μL. For this purpose, 10 μL of Red Mix solution, 5 μL of F and R primers, and 8 μL of double distilled water were added to 2 μL of the extracted DNA sample. The PCR temperature conditions were as follows: Following PCR, the products were electrophoresed (Figure 1).

Electrophoresis results of the polymerase chain reaction products related to the LRSAM1 gene with a length of 300 bp
The ABI 3130XI sequencer and Chromas software were used to confirm the variant. Variants were cross-referenced in ENSEMBL and Blast NCBI databases. Finally, Polyphen-2, Mutation Taster, Sift, and Predict SNP software were used to predict the potential pathogenic effects of the observed mutation.
3. Results
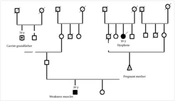
In this research, a family from Khuzestan Province that had a child with CMT disease was investigated. After genetic counseling and checking the family's history, the obtained variants were investigated (Figure 2).
.")
Family tree E-104 related to the LRSAM1 gene (nuclear family of a father, a pregnant mother who is her third child and a daughter and is in the 20th week of pregnancy, a daughter and a son, 20 years old, with Charcot Marie-Tooth has symptoms of muscle weakness in the arms and legs, as well as curvature of the spine).
After performing primary, secondary, and tertiary analyses on the data obtained from WES and based on the comparison of the resulting variants with the candidate gene for bone genetic disorders, a deleterious change in the form of NM_001005373.4:c.1985C>T (NC_000009.12:127501081:C:T), located on the locus q33.3-q34.11 of chromosome 9, was identified. This change could be caused by a homozygous or heterozygous mutation in the LRSAM1 gene (610933) on chromosome 9q33. According to NGS results, a homozygous mutation was found in the patient's family related to the LRSAM1 gene. Sequencing of coding exons showed that this mutation, NM_001005373.4:c.1985C>T, is located in exon 11, which has led to a change in codon translation and, ultimately, a change in the coding protein. Based on data from databases such as OMIM, the identified mutation was found to cause bone genetic disorders. The sequence results in all samples are similar in the region and codon number, and the only factor causing heterozygosity or homozygosity of individuals is the difference in the type of nucleotide in codon number 1985. The wild gene sequence has a C nucleotide in codon number 1985, whereas the variant gene sequence has a T nucleotide in codon number 1985. If both haplotypes related to this allele (allelic site in the maternal chromosome and allelic site in the paternal chromosome) have a C nucleotide in codon number 1985, the genotype is homozygous C (wild). If both loci have a T nucleotide, the genotype is homozygous T (homozygous and affected variant), and if one of the haplotypes contains a C nucleotide and the other contains a T nucleotide, the genotype is considered heterozygous CT for this variant (carrier). This interpretation is based solely on the exome genetic sequence, and the clinical interpretation related to the variant includes more components. Therefore, concerning the LRSAM1 variant, the nuclear family consists of heterozygous parents (carrier parents), 1 homozygous child (afflicted), and a completely healthy fetus that does not carry any defective paternal or maternal alleles. The detected mutation and the results of Sanger sequencing showed the different genotypes of the LRSAM1 variant in the parents (heterozygous) and the affected child (homozygous; Figure 3).

Sequencing results of parents with heterozygous genotype and affected child with homozygous genotype. The sequence results in all samples are similar in the area and codon number, and the only factor causing heterozygosity or homozygosity of individuals is the difference in the type of nucleotide in codon number 1985.
The probability of the variant being pathogenic was assessed using the POLYPHEN2 and SIFT sites, yielding results of 0.963 and 0.001, respectively. This variant lacks an ID and is not documented on various sites (CLINVAR, EXAC, HGMD, FRANKLIN), signifying its novelty.
4. Discussion
Charcot-Marie-Tooth disease, also known as hereditary motor and sensory neuropathy (HMSN), is clinically and genetically similar to hereditary neuropathy prone to pressure palsy (HNPP) and distal hereditary motor neuropathies (dHMN). Additionally, this disease is also referred to as distal spinal muscular atrophy (DSMA) (11). Epidemiological studies have demonstrated highly variable prevalence rates in different countries (12). Charcot-Marie-Tooth disease occurs due to the dysfunction of lower motor neurons and sensory neurons in the dorsal root ganglia or their lining glial cells (Schwann cells). The typical onset of CMT is during the first or second decade of life, although it can also manifest in infancy or in old age. Patients commonly experience slowly progressive distal muscle weakness and atrophy, often beginning in the legs and lower legs (13). The use of next-generation sequencing allows the simultaneous examination of multiple genes, distinguishing it from the Sanger sequencing method (14). In a specific study, the application of WES enabled the exploration of a gene panel encompassing 288 genes linked to sensory-motor disorders. This investigation led to the identification of the mutation NM_001005373.4:c.1985C>T:p.S662F in the LRSAM1 gene, reported for the first time. Analysis of the sequence in this region revealed a conversion of nucleotide C to T, resulting in a consequential mutation at codon 1985, leading to a termination codon. In 2019, Yoshimura et al. investigated new mutations in a statistical population of 301 people in Japan. From 40 genes, they identified pathogenic or possibly pathogenic variants in 301 cases (30%). GJB1 (n = 66) at 21.9%, MFN2 (n = 66) at 21.9%, and MPZ (n = 51) at 16.9% were the most common causative genes. In demyelinating CMT, variants were detected in 45.7% of cases, with GJB1 (40.3%), MPZ (27.1%), PMP22 point mutation (6.2%), and NEFL (7.7%) identified as the most common causes. Axonal CMT yielded a relatively lower detection rate at 22.9%. Their results indicated that the first decade of life is the most common period of onset of the disease, and the diagnosis of early CMT cases necessitates molecular confirmation (15). Furthermore, in 2023, Kontogeorgiou and colleagues conducted a genetic screening of 60 Greek patients using the WES technique. In their review, 20 cases were categorized as pathogenic or possibly pathogenic in the heterozygous state, encompassing 33.3% of patients. Among these, 14 were identified as pathogenic/probably pathogenic, and 6 were classified as pathogenic based on the ACMG classification after in silico evaluation. The most common genes involved included GJB1 (11.7%), MPZ (5%), and MFN2 (5%), followed by DNM2 (3.3%) and LRSAM1 (3.3%). Single cases with mutations in BSCL2, HSPB1, and GDAP1 were also identified (16). The research implies that the use of new techniques will notably advance the understanding of genes and mutations associated with this disease while also impacting the cost of diagnosis. Hence, this study endeavors to identify new mutations in the LRSAM1 gene responsible for bone genetic disorders within the affected family using advanced and effective techniques. It is evident that identifying genes and the resulting mutations pertinent to bone disease will significantly aid in elucidating the genetic complexities of this condition, subsequently improving prevention and counseling methods.
4.1. Conclusions
The results of this research demonstrate the significant potential of the WES method in discovering new genes and mutations, particularly among families affected by bone-related issues. Conducting further investigations involving a larger number of families could facilitate a better understanding of the prevalence and types of mutations associated with the referenced disease in Iran. This, in turn, could lead to the design of a specialized panel aimed at diagnosis and prevention, providing substantial assistance in averting new cases in families and contributing to the overall improvement of societal health.
