1. Background
Fabry disease (FD, OMIM #301500) is caused by the progressive accumulation of globotriaosylceramide (Gb3) and other glycosphingolipids, particularly in the plasma and in endothelial and smooth muscle vascular cells, cardiomyocytes, renal epithelial cells and cells of the autonomic nervous system, as a result of impaired activity of the lysosomal hydrolase, alpha-galactosidase (αGal, EC 3.2.1.22) (1, 2).
The human gene for αGal (GLA) is located at Xq22.1 and it comprises 7 exons which extend for 12,463 base pairs (1), with all their intron-exon splice junctions conforming to the GT/AT consensus sequence. The GLA gene is transcribed into a 1,418 nucleotide mRNA, containing a 5’-untranslated region (5’UTR) with 110 nucleotides but noticeably lacking a 3’-untranslated region. Its mRNA is translated into a 429 amino acid polypeptide, including a 31 residue signal peptide. The precursor polypeptide is processed to a 370 amino acid glycoprotein that associates in homodimers to form the mature enzyme (3). The human GLA gene additionally contains a cryptic exon in intron 4, which is variably expressed at trace to low levels (< 1% - 10%) in different tissues (4), leading to the in-frame insertion of 57 nucleotides in the non-canonical αGal transcript (c.639ins + 57), between exons 4 and 5. This latter GLA mRNA has a premature termination codon (PTC) just 10 codons downstream from exon 4 and its translation product is a non-functional, truncated polypeptide with 222 amino acids (4).
In males, who are hemizygous for X-linked genes, FD can be readily diagnosed by measuring αGal activity in cells, tissues or body fluids, most commonly in the plasma or in leukocytes (1, 2). In affected males, the severity of the disease phenotype and the earlier age of onset of its clinical manifestations are inversely correlated with the αGal residual enzyme activity (REA) measured in vitro (5): the childhood-onset, multi-systemic “classic” FD phenotype is typically observed in patients with REA < 1% - 5% of normal, while those with higher REA levels have later-onset, mostly cardiac complications of FD. Contrastingly, in affected females, as a consequence of the functional inactivation of one of the X chromosomes in early embryonic development (6), the measured REA activity reflects more the proportion of αGal deficient cells in the relevant tissues, rather than the intrinsic biochemical severity of the causative GLA mutation (7). For this reason, females who are heterozygous for pathogenic GLA mutations frequently show normal αGal activities and the gold standard for the diagnosis of FD in females is GLA genotyping (1, 2).
More than 750 different pathogenic GLA mutations are reported in “The Human Gene Mutation Database®” (HGMD®) [www.hgmd.cf.ac.uk/ac/gene.php?gene=GLA, last accessed on October 1, 2015], most of them being point mutations located within the coding sequences or in their flanking intronic regions, the latter typically resulting in exon skipping or misreading of the splice site. A further, peculiar type of splicing defect identified in FD patients (4, 8) is the overexpression of the non-canonical c.639ins + 57 transcript, in association with two deep intronic single nucleotide variants in intron 4, allegedly mediated by exonic splicing enhancer (ESE)-related mechanisms.
The molecular diagnosis of FD is usually achieved by Sanger sequencing of all the GLA exons and corresponding intronic boundaries in polymerase chain reaction (PCR) products amplified from genomic DNA samples. To improve the turnaround time and lower the cost of the molecular diagnosis of FD in the context of the PORTYSTROKE study (9), a large case-finding study among incident Portuguese patients with premature stroke we have developed a one-step reverse transcription PCR (RT-PCR)-based protocol for GLA genotyping by cDNA sequencing, which has since been used in our laboratory as the first-tier screening/diagnostic test for FD.
2. Objectives
Herein, we aim to report (i) that more than 20% of the patients with cerebrovascular disease (CVD) and/or left ventricular hypertrophy (LVH) referred for FD screening showed alternatively spliced transcripts of GLA cDNA involving exon 3; (ii) that such non-canonical transcripts are physiologically expressed at trace levels in healthy individuals; and (iii) that their expression in leukocytes markedly increases in blood samples kept at room temperature for 48 hours before RNA extraction.
3. Patients and Methods
3.1. Subjects, Blood Sampling and Baseline Laboratory Processing
Genomic DNA and total RNA specimens of patients prospectively enrolled in the PORTYSTROKE study, or of patients with CVD and/or LVH who had been referred to our laboratory for GLA genotyping for FD screening, were obtained from peripheral blood leukocytes, as described elsewhere (9). Most of the blood samples were mailed from other hospitals but were processed in our laboratory within 24 - 72 hours of collection.
Controls were 20 healthy men and women, selected among volunteer medical students and healthcare workers, specifically to match the age range of the PORTYSTROKE cohort. Two distinct venous blood specimens were collected on-site from each of the control subjects, at least one month apart. The first specimen was immediately processed for RNA extraction. The second specimen was collected into two tubes, one of which used for immediate RNA extraction, the other for RNA extraction following 48 hours standing at room temperature.
Total cellular RNA was extracted using either the RNeasy Mini Kit (Qiagen; Hilden, Germany) or the GF-1 Total RNA Extraction Kit (Vivantis Technologies; Selangor, Malaysia), according to the manufacturer’s instructions.
Informed consent was obtained from each patient and control included in the study and the study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki.
3.2. Molecular Genetic Analyses
Two partially overlapping amplicons, together comprising the GLA cDNA sequence from nucleotide position 22 through 1393, were synthesised by one-step RT-PCR. This ensured the mutational scanning of the entire αGal coding region except for the last two codons, where no pathogenic mutations have so far been reported to HGMD®. To avoid amplification of residual genomic DNA, the one-step RT-PCR primers were designed to pair with αGal mRNA sequences overlapping GLA exons 4 - 5 and exons 3 - 4, respectively (9, 10). Therefore, each of the final cDNA one-step RT-PCR products included a common sequence in their respective 3’ and 5’ terminals, corresponding to a part of exon 4.
To confirm the presence of the non-canonical transcripts, the following second-tier experiments were carried out in few selected patients: (i) two-step RT-PCR with the SuperScript® III First-Strand Synthesis System (Invitrogene, Life Technologies; Carlsbad, CA, U.S.A.), using the previously described reverse primers (9, 10) as gene-specific primers for cDNA synthesis, followed by PCR amplification with Platinum®Taq DNA Polymerase; (ii) one-step RT-PCR with a different set of primer pairs (Table 1) amplifying the GLA mRNA in three overlapping amplicons, with an overall sequence coverage similar to the standard protocol; (iii) one-step RT-PCR with primers specially designed to only anneal with the most frequent alternatively spliced transcript (Table 1). Finally, a second blood sample for RNA extraction was collected in a few selected patients, to check whether the GLA cDNA transcript profiles observed at baseline were persistently expressed, using the same laboratory protocol.
Table 1.
Primer Pair Sequences Used for One-Step RT-PCR Amplification Confirmatory Experimentsa
| Primer Code | Primer Oligonucleotide Sequence | Primer Coordinates |
|---|---|---|
| RTGLA 1F | 5’-ATAAGTCATCGGTGATTGGTC-3’ | 22 - 42 |
| RTGLA 3’4R | 5’-GTGCTTATAAC/CATCTGCCAA-3’ | 668 - 648 |
| RTGLA 2’3F | 5’-GCTAGCTAATTAT/GTTCACAGC-3’ | 467 - 488 |
| RTGLA 5’6R | 5’-GCCAATCACTAA/CATATCTGG-3’ | 923 - 903 |
| RTGLA 4’5F | 5’-CTTTCAAAAG/CCCAATTATACAG-3’ | 740 - 762 |
| RTGLA 7R | 5’-GTAAGTCTTTTAATGACATCTGC-3’ | 1395 - 1373 |
| RTGLA 3F | 5’-CTTCCCTGGGAGTTTTGGATA-3’ | 542 - 562 |
| RTGLA-62R b | 5’-GTGCTTATAAC/CAGTCAGCAA-3’ | 668 - 658/595 - 586 |
aPrimer coordinates are according to GenBank®. GLA cDNA nucleotide sequence version NM_000169.2 (National Center for Biotechnology Information (NCBI); Bethesda, MD, U.S.A.; www.ncbi.nlm.nih.gov/nuccore/NM_000169.2).
bThe last primer pair was specially designed to synthesise the alternatively spliced transcript.
To screen for splice site mutations in exon 3, its coding sequence and adjacent intronic boundaries were PCR-amplified from genomic DNA samples in all cases where GLA cDNA splicing isoforms of exon 3 were identified, using the primers and PCR conditions described elsewhere (10).
Moreover, in a few selected cases, the complete sequence of GLA exons 2, 3 and 4, and of introns 2 and 3, were amplified by long-range PCR (LR-PCR) in two large amplicons, respectively containing the < exon 2 - intron 2- exon 3 > and the < exon 3- intron 3 -exon 4 >. The primers and amplification conditions used for LR-PCR are summarised in Table 2.
| Primer Code | Primer Oligonucleotide Sequence | Primer Coordinates |
|---|---|---|
| GLA2-F | 5’-GGGAGGTACCTAAGTGTTCATTTA-3’ | 5035 - 5058 |
| GLA3-R | 5’-TGGCCTCAAAGTTCTTTCCTTTGTGGC-3’ | 7497 - 7471 |
| GLA3-F | 5’-ACCTGGTGAAGTAACCTTGTC-3’ | 7159 - 7179 |
| GLA4-R | 5’-GGGGAAGACACAAGGATGACT-3’ | 8617 - 8597 |
| Int2-5309F | 5’-TAAGCACTTCTGTACAGAAGCTTG -3’ | 5309 - 5332 |
| Int2-5656F | 5’-AGAAACAGCCCTCATGACAC-3’ | 5656 - 5675 |
| Int2-5712R | 5’-CCTGATAAGCCTTACAATTC-3’ | 5741 - 5712 |
| Int2-6073F | 5’-GTAATAGCTCTTGAGGCCCAT-3’ | 6073 - 6093 |
| Int2-6191R | 5’-AGTACCATTGCAATAGCTTCCT-3’ | 6212 - 6191 |
| Int2-6846R | 5’-TCCTAGCAAGACCTCTTAGC-3’ | 6865 - 6846 |
aPrimer coordinates according to GenBank®. GLA genomic DNA nucleotide sequence version X14448.1 (National Center for Biotechnology Information (NCBI); Bethesda, MD, U.S.A.; www.ncbi.nlm.nih.gov/nuccore/x14448.1).
bThe LR-PCR protocols consisted of an initial denaturation step at 94°C for 5 minutes, followed by 10 cycles of denaturation, annealing and elongation, respectively at 94°C for 15 seconds, 55°C (Gla2-F and Gla3-R) or 60°C (Gla3-F and Gla4-R) for 30 seconds and 68°C for 3 minutes, followed by 20 identical cycles with 20 extra seconds of elongation for each successive cycle and a terminal elongation step at 72°C for 7 minutes. Each 20 µL of the individual LR-PCR reaction mixture contained 2 µL of template DNA (100 ng/µL), 2 µL of the Expand Long Template Buffer 1 with 1.75 mM MgCl 1 µl of a 10 mM dNTP mix (100 mM dNTP Set; Invitrogen, Life Technologies; Carlsbad, CA, U.S.A.), 1 µl of the forward and reverse set of primers at 10 pmol/µl, and 0.3 µl of the Expand Long Template enzyme mix (5 U/µl; Roche; Mannheim, Germany).
The PCR, RT-PCR and LR-PCR products were checked on 1.5% agarose gel electrophoresis, and subsequently purified and automatically sequenced in forward and reverse directions, either in a 3100 or in a 3500 Genetic Analyzer (Applied Biosystems®, Life Technologies; Carlsbad, CA, U.S.A.), using standard laboratory methods. The primers used for the sequencing reactions were the same used for PCR, RT-PCR and LR-PCR; however, due to the large size and repetitive nature of GLA intron 2, specific primers had to be additionally designed to sequence that region (Table 2).
3.3. Alpha-Galactosidase Enzyme Activity Assay
In a subset of non-selected, consecutively enrolled stroke patients, the αGal enzyme activity was assayed in dried blood spots (DBS), according to published protocols (11).
3.4. GLA Sequence References
The following reference sequences of the nucleotide database of the National Center for Biotechnology Information (NCBI) (National Library of Medicine; Bethesda, MD, U.S.A.) were used as reference for the GLA genomic and mRNA/cDNA sequences, respectively: (i) X14448.1 [http://www.ncbi.nlm.nih.gov/nuccore/X14448.1]; (ii) NM_000169.2 [http://www.ncbi.nlm.nih.gov/nuccore/NM_000169.2].
GLA sequence variants have been described according to the recommendations of the human genome variation society (HGVS) (University of Melbourne; Melbourne, Australia) [http://www.hgvs.org/mutnomen/recs.html]. The A nucleotide in the ATG translation initiation codon was assigned position + 1 of the coding sequence. Single nucleotide polymorphisms (SNPs) and small deletion polymorphisms are reported according to their reference identification number (rs#) at the NCBI database of short genetic variations (dbSNP) [http://www.ncbi.nlm.nih.gov/SNP/].
3.5. Bioinformatic Analyses
The bioinformatic tools Berkeley Drosophila Genome Project Splice Site Prediction by Neural Network (BDGP) [http://www.fruitfly.org/seq_tools/splice.html] and NetGene2, a web server producing neural network predictions of splice sites in human DNA [http://www.cbs.dtu.dk/services/NetGene2/], were used to scan the wildtype GLA < exon 2 -intron 2 -exon 3 - intron 3 - exon4 > region for possible cryptic or low efficiency splice sites, and to predict the effects upon the normal recognition of exon 3 splicing sites, of sequence variants eventually identified in the adjacent introns. The ESEfinder software [http://rulai.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi] was used to search for putative cis-acting exonic splicing enhancers (ESE) in the same genomic regions.
3.6. Statistical Analyses
All statistical analyses were performed with GraphPad Prism, version 5.0 (GraphPad Software; La Jolla, CA, U.S.A.).
4. Results
4.1. Non-Canonical GLA cDNA Exon 3 Transcripts Were Identified in About One-Fourth of Patients With Cerebrovascular Disease or Left Ventricular Hypertrophy
Among 482 patients (247 males and 235 females) with CVD or unexplained LVH, 367 (76.1%) showed only the canonical GLA cDNA transcript on the agarose gel electrophoresis, while the remaining 115 (23.9%) presented one or more alternatively spliced transcripts, either with or without the canonical transcript (Table 3). Notably, a transcript lacking the last 62 nucleotides of exon 3 (c.del486-547), was identified in 113 cases (98.3%) and 4 patients had a transcript lacking all of the 178 nucleotides of exon 3 (c.del370-547). The canonical and the alternatively spliced GLA transcripts corresponding to relevant cDNA sequences are illustrated in Figure 1.
Table 3.
Number of Patients Presenting the Canonical and the Non-Canonical GLA Transcripts
| Males | Females | |
|---|---|---|
| Wildtype/c.del486-547 | 39 | 52 |
| Wildtype/c.del370-547 | 1 | 1 |
| c.del486-547 | 12 | 8 |
| Wildtype/c.del486-547/c.del370-547 | 1 | 1 |

Figure 1.
Electropherograms (ABI PRISM 3100 Genetic Analyzer®) of A, Canonical exon 3 sequence; B, Exon 3 and 4 boundary sequence resulting from the splicing of the last 62 nucleotides of exon 3; C, Canonical exon 2 and 3 boundary sequence; D, Exon 2 and 4 boundary sequence resulting from the deletion of the entire exon 3. In boxes wildtype codons; Shadowed-newly formed codons resulting from the deletion, leading to translational frameshifting and premature termination.
The presence of the non-canonical transcripts was documented in all cases selected to perform the confirmatory second-tier one-step and two-step RT-PCR analyses. Of note, in 8 of the 10 cases who had shown only the canonical transcript in the original one-step RT-PCR analysis, the c.del486-547 could be detected in minor amounts, using the primers specifically designed to anneal with this transcript.
In the few patients where a confirmatory one-step RT-PCR analysis was subsequently carried out on RNA specimens extracted from a second blood sample, the GLA cDNA transcript profiles observed in each subject did not match with the former in most cases.
4.2. The Generation of the Non-Canonical GLA cDNA Exon 3 Transcripts Could not be Explained by Splice Site or Deep Intronic Sequence Variants
Most patients who presented any of the two non-canonical GLA exon 3 transcripts had the wildtype genomic DNA sequence of exon 3 and of its adjacent intronic boundaries. The c.370-81-370-77delCAGCC deletion polymorphism in intron 2 (rs5903184) [http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi? rs = 5903184] was identified in about 10% of the patients; however, the prevalence of this variant did not differ among individuals presenting only the canonical or any of the non-canonical GLA cDNA transcripts, and was similar to the prevalence observed in the Portuguese population [Ferreira S et al. Frequencies of allelic variants of the GLA gene in Portuguese healthy individuals and stroke patients. Abstract in Clinical Therapeutics 2009; 31 Suppl. A: S36].
In eight selected cases, including two subjects who had only the canonical GLA transcript, four who had only the c.del486-547 transcript, one who had both the canonical and the c.del486-547 transcript, and one who had both the canonical and the c.del370-547 transcript LR-PCR was used to scan for deep intronic mutations in introns 2 and 3, as well as for splice site mutations in exons 2, 3 and 4. Seven of those individuals had the wildtype gene sequence but the patient presenting both the canonical and the c.del486-547 GLA transcripts carried the g.7636C > T/c.547 + 190C > T SNP in intron 3 (rs41311559) [http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi? rs = 41311559].
4.3. Bioinformatic Analyses
According to bioinformatic analyses, the wildtype donor splice site of GLA exon 3 is not robustly defined and the deep intronic c.547 + 190C > T SNP does not interfere with the splicing mechanism.
According to the in silico predictions of BDGP and NetGene2, the acceptor splice sites of GLA exons 3 and 4 are robustly defined, with scores or confidence values of 0.98 to 0.99. On the other hand, the donor splice sites of exons 2 and 3 are either unrecognized or only weakly defined (BDGP score for the exon 2 = 0.66; NetGene2 confidence value for exon 3 = 0.61). Notably, the ESEfinder software identifies a putative high-score ESE binding site for Serine/Arginine-rich protein 40 (SRp40) a protein involved in the recruitment of spliceosomal components (12) to the nucleotides c.480 through c.486 of GLA exon 3.
In silico analyses with BDGP, NetGene2 and ESEfinder showed that the c.547 + 190C > T SNP does not change the predicted splice site scores of exons 2, 3 and 4, and does not create or eliminate any ESE consensus motifs within the same region, as compared to the wildtype sequence.
4.4. The Whole Blood αGal Enzyme Activities in Patients With Non-Canonical GLA cDNA Transcripts Were Not Decreased
After the recurrent identification of alternatively spliced GLA cDNA transcripts in PORTYSTROKE patients, DBS in filter paper were prepared from the venous blood samples of 81 consecutively enrolled cases, to check whether the presence of non-canonical transcripts correlated with αGal enzyme deficiency in the whole blood assay. Remarkably, the distribution of the αGal enzyme activities of the 76 patients who showed only the canonical transcript, and of the remainder 5 patients, who presented the GLA c.del486-547 variant transcript, either alone (n = 3, including one female) or together with the normal transcript (n = 2) did not statistically differ (two-tailed t-test: P = 0.58) (Figure 2).

Figure 2.
Distribution of the Whole Blood αGal Activity Values in 81 Incident Young Stroke Patients
4.5. Effect of Delayed RNA Extraction on Observation of Non-Canonical GLA cDNA Transcripts
Non-canonical GLA cDNA transcripts involving exon 3 were also observed in healthy individuals when RNA extraction was delayed for 48 hours.
Using our standard one-step RT-PCR approach to identify GLA mutations, no alternatively spliced cDNA transcripts could be identified using agarose gel electrophoresis for any of the 40 control RNA samples that were extracted immediately upon blood collection. Contrastingly, the c.del486-547 non-canonical transcript was identified in 17 of the 20 healthy controls when their blood samples were left standing at room temperature for 48 hours before RNA extraction. In the remaining three cases, the presence of non-canonical exon 3 GLA cDNA transcripts could not be resolved with certainty either using agarose gel electrophoresis or in the sequencing analysis.
When the one-step RT-PCR analysis was carried out with primers specifically designed to anneal with the non-canonical c.del486-547 GLA cDNA, this transcript was detected in 7 of the 8 control RNA samples, including in all 4 that were obtained immediately after blood sampling.
5. Discussion
Alternative splicing is a post-transcriptional process in eukaryotic organisms by which multiple distinct mRNA transcripts are produced from a single gene (13). It has been estimated that up to 95% of human multi-exon protein coding genes undergo alternative splicing, often in a tissue- or developmental stage-specific manner (13-15), and in response to external stimuli as part of several signal transduction networks (13, 15). The generation of multiple distinct functional mRNA transcripts from a single gene, by one of the four major mechanisms of alternative splicing, is an important source of proteomic diversity, with critical roles in the control of developmental processes and in the dynamic regulation of the transcriptome (13, 15, 16). Alternative splicing may have contributed to functional innovation during the evolution of the eukaryotic genome (14). The splicing process is performed by the spliceosome, a ribonucleoprotein megaparticle that assembles around splice sites at each intron (13, 17). The mechanism of RNA splicing is highly complex, requiring multiple interactions between pre-mRNA, small nuclear ribonucleoproteins and a multitude of splicing factor proteins. Trans-acting factors, mainly RNA-binding proteins, modulate the activity of the spliceosome and of cis-acting RNA sequences, which include exonic and intronic splicing enhancers and silencers (13-15, 17). In addition to their putative role in constitutive splicing, these cis-regulatory elements are also involved in the regulation of alternative splicing. Up to one-third of alternatively spliced transcripts have been found to contain a PTC (14, 16). Since these aberrant transcripts are apparent targets of nonsense-mediated mRNA decay (NMD) (13, 16-18), the coupling of alternative splicing and NMD may be a pervasive means of regulating protein expression, a process that has been called regulated unproductive splicing and translation (RUST) (16). Mutations in regulatory sequences that affect alternative splicing are a widespread cause of human hereditary disease and cancer (13, 15). About 10% of all human pathogenic mutations identified in diagnostic molecular genetics laboratories are in canonical splice sites (19), but this estimate does not include mutations affecting splicing enhancers, silencers or trans-acting factors, which are much more difficult to recognise and may have been historically overlooked (20).
With our cDNA GLA genotyping protocol, using RNA extracted from peripheral blood leukocytes, we have identified two novel alternatively spliced transcripts affecting exon 3, at concentrations high enough to be detectable on agarose gels, one lacking the last 62 nucleotides (c.del486-547), the other missing the entire exon (c.del370-547). While the latter was uncommonly found, the c.del486-547 transcript was present in more than 20% of the GLA cDNA samples analysed in the PORTYSTROKE study. These alternatively spliced GLA transcripts occurred in the absence of any sequence variants that might affect the splicing mechanism.
GLA intron 3 is a phase 1 intron, intervening between the first and the last two nucleotides of the glycine encoding codon 182. The two non-canonical transcripts lead to translational frameshifting, generating PTCs respectively 2 and 9 codons downstream from the c.del486-547 and c.del370-547 deleted segments. Therefore, these two alternatively spliced transcripts are most probably targeted to NMD (13, 16, 17) and, when overexpressed, might be the cause of reduced αGal enzyme activity. Although the distributions of the DBS αGal activity levels in stroke patients presenting the c.del486-547 GLA transcript or exclusively with the canonical transcript were not statistically different, it should be borne in mind that the DBS αGal assay correlates better with the plasma than with the leukocyte αGal assay (21) and, therefore, may not be the most reliable gauge of the GLA mRNA expression in leukocytes.
Studies in healthy subjects, carried out to determine whether the non-canonical GLA transcripts were physiologically expressed and whether the holding time of the blood samples before RNA extraction might affect their expression, indicated that, at least in leukocytes, the c.del486-547 transcript is constitutively expressed at trace levels, which are only detectable with specific RT-PCR amplification, but become visible on agarose gels when the RNA is extracted from leukocytes that were left standing in the whole blood sample, at room temperature, for 48 hours. These findings suggest that, under those circumstances, the genetic metabolic environment in leukocytes favors the accumulation of the non-canonical c.del486-547 transcript, either by increased production or decreased degradation.
According to the in silico predictions, the donor splice sites of exons 2 and 3 are relatively weaker in comparison to the acceptor splice sites of GLA exons 3 and 4. Although the first two nucleotides that are excluded in the alternatively spliced sequence of exon 3 are guanines, therefore not corresponding to a canonical donor splicing consensus motif, the terminal sequence of exon 3 remaining in the c.del486-547 transcript contains a putative high-score ESE binding site for Serine/Arginine-rich protein 40 (SRp40), which is one of the key proteins involved in the recruitment of spliceosomal components (12). These conditions would favor the alternative recognition of a non-canonical donor splice site within exon 3 that might explain the generation of the GLA c.del486-547 transcript.
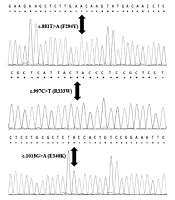
The overexpression of a cryptic exon in intron 4, leading to extremely unbalanced ratios of the c.639ins + 57 over the wildtype GLA transcript, has already been identified as the underlying cause of deficient αGal activity in (i) patients with the cardiac variant of FD associated with carrying the mid-intronic g.9331G > A/c.640-801G > A SNP (rs199473684) [http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi? rs = 199473684] originally reported as c.639 + 919G > A (4) mapping 4 nucleotides before the 3’ end of the cryptic exon, who exhibited REA at ≈10% of the normal level (4); and (ii) in a patient with the classic phenotype of FD and who exhibited REA at ≈1% of the normal level (8), associated with the novel mid-intronic variant g.9273C > T/c.640-859C > T originally reported as c.861-5C > T (8) mapping 5 nucleotides upstream from the beginning of the cryptic exon.
The hypothesis that the non-canonical c.639ins + 57 GLA transcript is overexpressed in patients carrying the g.9331G > A SNP due to increased recognition of the alternative splicing by an A/C-rich enhancer-type ESE (4), generated by the G > A transition, has been disputed (22) since that SNP occurs at a non-consensus site of the cryptic donor splice site and there are no functional ESE motifs (23) within the surrounding intronic region. Bioinformatic analyses of the g.9273C > T variant indicated that this transition does not significantly change the predicted acceptor site score, in comparison to the wildtype sequence, and that the highly predominant expression of the c.639ins + 57 transcript might be explained by the creation of a novel ESE (8).
The alternatively spliced c.639ins + 57 transcript was not identified in any of our study patients, suggesting that the c.640-859C > T and the c.640-801G > A (rs199473684) intronic variants are uncommon in the Portuguese population.
As a conclusion, we hypothesise that the production of alternatively spliced GLA transcripts containing PTCs might be physiologically involved in the post-transcriptional regulation of GLA gene expression, and that its dysregulated overexpression, especially if limited to specific cells or tissues, might be the cause of Gb3 storage in affected tissues with no pathogenic mutation identified in the GLA gene, like the recently described Gb3-associated cardiomyopathy (24). Elucidation of the mechanism underlying the production of these abnormal GLA transcripts, and of their biological consequences, warrants further investigation as they may contribute important new data to the understanding of the molecular pathology of FD and Gb3-related disorders.