


1. Introduction
Fraser syndrome (FS) is a rare genetic disorder with an autosomal recessive inheritance that has been reported in just over 250 cases so far (1). This syndrome affects an estimated 0.04 in 10,000 live births and occurs in approximately 1 case per 10,000 stillbirths (2).
Characteristic features of FS include cryptophthalmos, syndactyly, and abnormal genitalia, which may be coupled with other abnormalities in the urinary tract, ear, nose, and larynx (3). Cryptophthalmos, which is characterized by the lack of palpebral fissures in conjunction with severe microphthalmia or anophthalmia and absent or malformed lachrymal ducts, has been reported in 85% - 93% of cases and partial cutaneous syndactyly is present in more than half of cases (3). Several other malformations that have been described in this syndrome include nasal malformations (especially a broad, flat, or depressed nasal bridge), malformations of the ears (e.g. distorted and/or low set ears, microtia, and atresia or stenosis of the external auditory meatus), malformations of the respiratory system such as laryngeal stenosis or atresia, orofacial clefting, musculoskeletal anomalies, gastrointestinal malformations (most commonly imperforated anus), renal, cerebral and cardiac malformations, thymic abnormalities and developmental delay and psychomotor retardation (4).
The FRAS1, FREM2, and GRIP1 genes are located on chromosomes 4q21, 13q13, and 12q14, respectively. Heterozygous or homozygous mutations in these three genes have been found to cause FS (5).
Twenty-five percent of infants with FS are stillborn and it is estimated one-fifth of the affected newborns die before their first birthday because of renal or laryngeal anomalies (3). This manuscript is a case report of a Fraser Syndrome and a review of the literature related to the renal malformations associated with FS.
2. Case Presentation
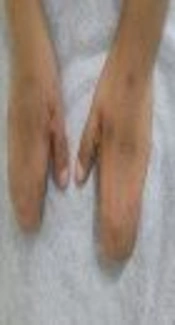
A 6-year-old girl with multiple congenital anomalies attended the provincial clinical genetic counseling center, Zahedan, Iran. She was a result of a full term normal vaginal delivery. She was born to consanguineous parents (i.e. first cousins) with no history of genetic disorders. In the physical examination, we found total unilateral cryptophthalmos (left side), low hairline, depressed nasal bridge, small posterior rotated ears (Figure 1A), a low set umbilicus, enlarged clitoris, hypoplastic labia majora, and cutaneous syndactyly of both hands and left foot (Figure 1B and C). She had a history of difficult intubation because of laryngeal stenosis. Ultrasonography of the abdomen showed unilateral renal agenesis (left side) without agenesis of the ureter. In the molecular analysis, mutations were reported in the FRAS1 gene Exon58:C. 8698G-T (homozygous). The parents were heterozygous for the same mutation. In the following pregnancy, the patient’s mother gave birth to an apparently normal baby. The baby was also a carrier for the above-mentioned mutation.

A, complete cryptophthalmos and a flat nose; B, bilateral cutaneous syndactyly, hands; C, unilateral cutaneous syndactyly, left foot.
3. Discussion
According to the diagnostic criteria proposed by Thomas et al. in 1986, the clinical manifestations in our case suggested a diagnosis of FS (6). The main characteristics considered as major criteria include cryptophthalmos, syndactyly, anomalous genitalia, and having a brother or sister with cryptophthalmos syndrome. A series of symptoms that comprise the minor criteria include congenital abnormalities of ears, nose, larynx, and skeleton, cleft lip, renal agenesis, umbilical hernia, and mental retardation. For a diagnosis of FS, two major criteria and one minor criterion or one major and at least four minor criteria should be present. In our case, unilateral cryptophthalmos, syndactyly, and abnormal genitalia (enlarged clitoris, hypoplastic labia majora) were the major criteria. Malformations in the ears, nose, laryngeal stenosis, and unilateral renal agenesis comprised the minor criteria.
Mutations in the FRAS1 gene were found in this FS case and the parents were heterozygous for the same mutations. FS can be a result of mutations in FRAS1 and FREM2 genes and about half of the FS cases have been found to link to FRAS1 gene mutations (7). Thus far, 26 mutations (mostly truncating changes) have been identified in FRAS1 and FREM2 genes. These mutations vary from nucleotide substitutions, splicing defects, and large insertions to small deletions/insertions (1).
The FRAS1 and FREM2 genes produce the FRAS1 and FREM2 proteins, respectively. These groups of proteins create the FRAS/FREM complex that comprises the extracellular matrix proteins. The FRAS/FREM complex found in basement membranes plays an important role in maintaining the structural integrity of tissues. This complex is also fundamental to organ morphogenesis as it mediates the epithelial-mesenchymal interactions (8). The findings from mouse models and developmental defects reported in FS suggest that complete or near complete lack of function or loss of any of these proteins may give rise to the disintegration of the abovementioned protein complex (9). Deficiencies in the FRAS-FREM complex can also result in clinically overlapping but distinctive and less severe group of diseases including Manitoba-oculo-tricho-anal (MOTA) and anorectal malformations (BNAR) syndromes, bifid nose, and renal agenesis (10).
However, a molecular study of 33 families with FS showed that FRAS1 and FREM2 gene mutations were present in only 43% of the investigated cases. This finding proposes that FS could be caused by mutations in genes other than FRAS1 and FREM2 (11). During the early stages of the embryonic development, GRIP1 gene activity is necessary for normal cell-matrix interactions. This fact has been supported by observations in the mouse models showing that inactivation of GRIP1 gene can result in FS-like defects (8). The GRIP1 gene produces the GRIP1 protein. This protein plays an important role in the formation of the FRAS/FREM complex by meditating the process for FRAS1 and FREM2 to reach the proper location of the cell (12).
Unilateral renal agenesis was observed in our FS case. A wide range of renal malformations has been found to be associated with FS. Bilateral or unilateral renal agenesis that may be or may not be coupled with ureteral agenesis has been reported in 23% and 22% of the FS cases, respectively (4). Other malformations frequently reported include absent, hypoplastic, or small bladder with or without urethra (17%) and cystic dysplasia of the kidneys (12%) (4). Agenesis of entire urinary apparatus has also been reported. Therefore, FS should be considered as a diagnosis in all cases of stillbirth that are coupled with renal agenesis.
Although the mechanism of renal malformations associated with FS is unclear, it has been hypothesized that the FRAS/FREM complex plays an important role in the proper development of organs and tissues, including the kidneys (13). Mutations in the genes encoding FRAS1, FREM1, and FREM2 have been reported to result in dermal-epidermal detachment in the plane of sublamina densa. Different classes of mouse bleb mutants that are the murine model of human FS have shown similar pathological changes characterized by embryonic skin blistering, cryptophthalmos, and renal agenesis (12). Branching ureteric bud epithelia has shown to be covered by the FRAS1 protein that is markedly upregulated in the nephron lineage after mesenchymal/epithelial transition (14). FRAS1 protein deficiency may give rise to defective interactions between the bud and mesenchyme, which in turn decreases the expression of key nephrogenic molecules. Furthermore, FRAS1 protein is thought to play a role in the formation of normal glomeruli (14). Moreover, single heterozygous missense mutations in FRAS1 gene have been linked to non-syndromic unilateral renal agenesis (1).
Isolated congenital malformations of the kidney and urinary tract are partially associated with some mutations in FRAS1, FREM2, GRIP1, FREM1, ITGA8, and GREM1 genes with a recessive inheritance. These findings further support the importance of the FRAS/FREM complex in the pathogenesis of renal malformations in FS (15).
Given the fact that the recurrence of FS is likely to occur in 25% of the siblings, prenatal diagnosis (PND) and genetic counseling should be considered for affected families (16). A combination of ultrasound and fetoscopy could be used for prenatal diagnosis of FS (16). Although oligohydramnios has been found to be the most frequent complication of FS during pregnancy, other complications such as anhydramnios, antepartum hemorrhage, vaginal bleeding, hydrops fetalis, nuchal edema, fetal ascites, intrauterine growth retardation, and hypoplasia/single umbilical artery have also been described to be associated with FS (4). It has been reported that 15% - 25% of the FS cases are a result of a consanguineous marriage, which also was the case in this report (4).
Considering the relatively high rates of fertility and inbreeding marriages in Sistan and Balochistan province, clinicians should pay special attention to considering a possibility of the diagnosis of FS based on clinical findings. They should recommend the identified FS cases a comprehensive follow-up and genetic counseling to their parents and PND services in future pregnancies.
