


1. Background
Deficiency of phenylalanine hydroxylase, an enzyme produced by the liver, leads to phenylketonuria (PKU), which is an autosomal recessive genetic disorder affecting the metabolism of phenylalanine. Phenylalanine (1) is converted to tyrosine by phenylalanine hydroxylase (PAH; EC 1.14.16.1) (1, 2). The defective enzyme leads to the accumulation of Phe in blood and body fluids of PKU patients (3) and can cause seizures, severe intellectual disability (mental retardation), and other diseases in untreated cases. The incidence of PKU is one in 3627 live births in nine of all Iranian populations, but it differs among others (4, 5).
Mutations in the PAH gene on chromosome 12q23.2 occur in 98% of PKU patients (6). So far, over 600 different mutations have been identified in PAH that are listed in databases of PAH and HGMD. A high percentage of them are point mutations arising from single base substitution (2, 6). PAH enzyme dysfunction mechanisms depend on the PAH gene mutation location and type (7). The major damaging effect on its structure and function is caused by improper folding resulting in decreased protein stability and causing degradation (8). Bioinformatics studies of nonsynonymous genetic variants effects could help identify more likely disease-causing variants. As experimental studies are expensive in nature (and due to the difficulty that it entails), several studies have used bioinformatics analysis to predict potentially damaging effects of nonsynonymous single nucleotide polymorphisms (SNPs) in humans (9). Jahantigh and Hosseinzadeh Colagar used in silico analysis to evaluate the biological function of rs6147172 and rs2267437 SNPs. They predicted the effects of this variation on the splicing of XRCC7 mRNA by using HSF3, Human Splicing Finder version 3, SplicePort, and SpliceAid 2 (10). Ernst et al. classified rare BRCA1/2 missense variants using four prediction tools, Align-GVGD, SIFT, PolyPhen-2, and MutationTaster2 (11). Macauda et al. used in silico analysis to evaluate the relationship between increased gene expression and variant alleles of four SNPs in linkage disequilibrium with ABCC2‐rs4148388 (12). Salimi et al. studied the association between systemic lupus erythematosus and ERα polymorphisms using in silico analysis (13).
2. Objectives
In the present study, an in silico analysis was designed to evaluate the pathogenicity rate of a novel variant Q375R (c.1124A>G) and three intron variants, IVS9 + 32insA, IVS11 + 163delC, and IVS12 + 30C>T (14).
3. Methods
3.1. Mutation Analysis
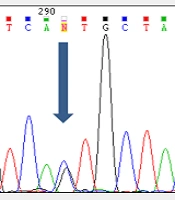
We had analyzed the phenylketonuria mutations in south-west Iran in a previous study. In summary, 40 unrelated hyperphenylalaninemia patients were selected. Blood samples were collected from all cases (2.5 - 3 mL). Diatom Prep 100 kit was used to extract DNA. PAH mutations in 6, 7, and 10 - 12 exons and the adjacent flanking region were studied using the polymerase chain reaction (PCR) with manually designed primers. After the purification of PCR products, an automated sequencing was performed using applied biosystems 3730 DNA analyzer (Macrogen, Seoul, Korea). The sequence alignment was evaluated using the National Center for Biotechnology Information (NCBI) BLAST tool. Totally, 13 different mutations were identified including six missense mutations, three nonsense mutations, three splice site mutations, and one deletion. Intronic regions of PAH contained three novel variants and seven various polymorphisms. The c.1124A>G (Q375R) alteration was a novel variant identified in exon 11. Three other new variants, IVS9 + 32insA, IVS11 + 163delC, and IVS12 + 30C>T, were detected in introns 10, 12, and 12, respectively (6).
3.2. Evolutionary Conservation Analysis
The Q375R variant was analyzed in silico. The evolutionary conservation of protein sequences was studied by PANTHER (protein analysis through evolutionary relationships) (15) and MutationTaster, a program using phastCons and phyloP to determine the given nucleotide conservation grade (16).
3.3. Analysis of Deleterious Variation
Damaging effects of single nucleotide variation c.1124A>G were analyzed using seven tools: PolyPhen-2 (polymorphism phenotyping v2), MutPred, SNAP2 (screening for non-acceptable polymorphisms 2), SNPs&GO, SIFT (sorting intolerant from tolerant), MutationTaster, and nsSNPAnalyzer. SIFT is a tool predicting the effects of amino acid substitution on protein function (deleterious or neutral). This software uses the conservation degree of amino acid residues and the physicochemical properties of amino acids (17). The PolyPhen-2 tool utilizes a combination of sequence and structure-based attributes and uses naive Bayesian classifier for the identification of an amino acid substitution and the effects of the mutations (18). MutPred predicts the probability of amino acid deleterious substitutions and the molecular cause of disease-related amino acid substitutions (19). The SNPs&GO method uses protein functional annotation to predict the SAPs (single amino acid polymorphisms) (20). SNAP only uses sequence-based computationally acquired information and potentially classifies all nsSNPs (non-synonymous coding SNPs) into neutral and non-neutral classes (21). Based on the alteration type, MutationTaster makes a choice between three distinct prediction models (aiming at either intronic alterations or silent synonymous) at alterations affecting a single amino acid or at alterations which cause changes in the sequence of the amino acid (16). The nsSNP Analyzer server is able to integrate several protein structure analyses and sequence alignments to identify nsSNPs associated with diseases. nsSNP Analyzer uses the sequence of a protein and the accompanying nsSNP to report if an nsSNP could be disease-associated or it is functionally neutral (22).
3.4. Prediction of Protein Structural Stability
I-Mutant 3.0, foldX, and Mustab were used to predict the effects of the altered amino acid on the stability of the protein (19, 23).
3.5. Structural Analysis
Structural analysis was performed using Swiss Pdb viewer 4.1.0 and phyre2, which is a web tool to predict and build 3D models (25). After that, the ligand binding sites were predicted using 3DLigandSite (26) and the structure was visualized by Molegro Virtual Docker 5.5.
3.6. In Silico Analysis of Non-Coding Variant
For understanding the effects of three novel non-coding variant sequences (IVS9 + 32insA, IVS11 + 163delC, and IVS12 + 30C>T) on RNA splicing, the variations were studied by MaxEntScan, FSPLICE, GeneSplicer, and human splicing finder.
4. Results
4.1. Variant Position
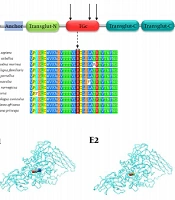
UniProt data were analyzed using Office to evaluate variant positions. Figure 1 shows the PAH gene, domains, and the position of the desired variants.

Figure 1.
The analysis of variant position showing PAH gene, ACT domain, and the variants
4.2. Conservation Evaluation of Variant Location
The results of MutationTaster showed phastCons values equal to 1, indicating a higher probability of nucleotide conservation. In addition, phyloP showed a score of 4.96 (the closer the value to 6, the higher the probability of conservation). Evolutionary analysis of coding SNP in the PANTER tool resulted in a NIC value of 4.402. A smaller value of NIC indicates a higher probability of not being conserved in the multiple sequence alignment for the variation.
4.3. Pathogenicity Prediction of Deleterious Variation
Q375R was predicted to be deleterious using four tools, SIFT (score 0.00), PolyPhen (PSIS 0.985), Mutpred (score 0.816), and MutationTaster. SNAP predicted Q375R to be intermediate for damaging with a score of 46, but nSSNP analyzer predicted Q375R to be a neutral variant.
4.4. Stability Prediction of the Novel Variant Q375R
The tools I-Mutant 3.0, Fold X, and Mustab evaluated the levels of stability in the protein. Fold X predicted that the mutation of Gln to Arg (position 375) results in a ddG of 0.83 kcal/mol. According to the result, the mutation moderately decreases the stability of the protein. The obtained results from other tools including Mustab (confidence of 83.04) and I-Mutant 3.0 (ddG of 0.06 kcal/mol, slight) also imply that the protein stability decreases upon Q375R variation.
According to the output of the SNP effect, TANGO predicted the new variant effects on the aggregation-prone regions of the protein sequence. For Q375R, dTANGO equals 0.4, meaning that the aggregation tendency of the protein is not affected by this mutation (Table 1).
Table 1.TANGO Regions in Both Wild Type and Variant Proteinsa
| Number | Start | End | Stretch | Score |
|---|---|---|---|---|
| Wild type | ||||
| 1 | 33 | 41 | AISLIFSL | 65.05 |
| 2 | 320 | 329 | LATIYWFTV | 88.72 |
| Mutant | ||||
| 1 | 33 | 41 | AISLIFSL | 65.07 |
| 2 | 320 | 329 | LATIYWFTV | 88.72 |
aStart, end, sequence, and score are given for TANGO regions.
The aggregation-prone regions predicted by TANGO were visualized in the protein structure (Figure 2). As an output of SNPeffects, Waltz predicted the new variant effects on amyloid-forming regions. For the Q375R variation, dWALTZ equals -537.22 which means this amino acid change decreases the amyloid propensity of the protein. Table 2 shows the differences between the aggregation profiles of the wild type and the mutant protein.
Table 2.WALTZ Regions in Variant and Wild Type Proteinsa
| Number | Start | End | Stretch | Score |
|---|---|---|---|---|
| Wild type | ||||
| 1 | 33 | 39 | AISLIF | 80.99 |
| 2 | 161 | 167 | ADIAYN | 75.41 |
| 3 | 201 | 207 | ACYEYN | 28.21 |
| 4 | 322 | 328 | TIYWFT | 6.38 |
| 5 | 372 | 378 | AIQNYT | 95.60 |
| Mutant | ||||
| 1 | 33 | 39 | AISLIF | 80.97 |
| 2 | 161 | 167 | ADIAYN | 75.41 |
| 3 | 201 | 207 | ACYEYN | 28.21 |
| 4 | 322 | 328 | TIYWFT | 6.37 |
| 5 | 372 | 378 | AIQNYT | 6.10 |
aStart, end, sequence, and score are given for WALTZ regions.

Figure 2.
The molecular visualization of TANGO aggregation-prone regions. TANGO aggregation-prone regions are shown as red-colored segments in the molecular image. The structural location of the variant residue is colored in yellow.
Chaperones are essential elements in controlling protein folding and degradation of misfolded proteins. The results of LIMBO for the prediction of chaperone binding site shows dLIMBO equal to -0.92, meaning that the chaperone-binding tendency of the protein is not affected by the new variant. Short segments are listed in the table for both wild type and newly varied proteins. The results show an increase in one of the stretches from position 385 to 393 (Table 3).
Table 3.LIMBO Regions in Varied and Wild Type Proteinsa
| Numbers | Start | End | Stretch | Score |
|---|---|---|---|---|
| Wild type | ||||
| 1 | 49 | 58 | KVLRLFEEN | 88.78 |
| 2 | 115 | 123 | DTVPWFPR | 100.00 |
| 3 | 187 | 195 | GTVFKTLK | 99.42 |
| 4 | 239 | 247 | FRLRPVAG | 40.37 |
| 5 | 326 | 334 | FTVEFGLC | 100.00 |
| 6 | 376 | 383 | YTVTEFQ | 85.08 |
| 7 | 397 | 405 | KVRNFFAAT | 11.52 |
| Mutant | ||||
| 1 | 49 | 58 | KVLRLFEEN | 88.78 |
| 2 | 115 | 123 | DTVPWFPR | 100.00 |
| 3 | 187 | 195 | GTVFKTLK | 99.42 |
| 4 | 239 | 247 | FRLRPVAG | 40.37 |
| 5 | 326 | 334 | FTVEFGLC | 100.00 |
| 6 | 376 | 383 | YTVTEFQ | 38.23 |
| 7 | 385 | 393 | YYVAESFN | 5.97 |
| 8 | 397 | 405 | KVRNFFAAT | 47.87 |
aStart, end, sequence, and score are shown for LIMBO regions.
4.5. Effect of Q375R on Protein Structure
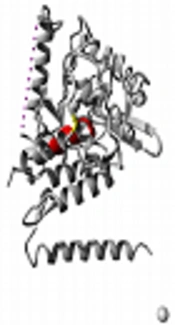
The overall 3D structure of PAH protein and the position of 375 amino acid were investigated using Swiss Pdb viewer 4.1.0 software (Figure 3). According to the phyre2 modeling and the result of 3DLigandSite, the HIS 285 and HIS 290 residues were predicted to be a part of the ligand-binding site in the wild type protein. For the varied protein, GLU 330 was present in PAH binding sites in addition to HIS 285 and HIS 290 (Figure 3).
")
Figure 3.
A, The 3D structure of PAH protein and the position of 375 amino acid; B, wild type protein; and C, mutant protein (Q375R)
4.6. Effect of Variations in the Intronic Region
Three novel non-coding variants, IVS9 + 32insA, IVS11 + 163delC, and IVS12 + 30C>T, were predicted as polymorphism by GeneSplicer, Human Splicing Finder, MaxEntScan, and FSPLICE. Analysis using these software packages showed that the variations had no significant effect on RNA splicing.
5. Discussion
PKU, an autosomal recessive genetic disorder with metabolic impairment, is mainly caused by mutations in the PAH gene. These mutations mostly result in deficient enzyme activity and subsequently cause hyperphenylalaninemia. They occur mainly in the exonic and flanking regions of the gene. The majority of these variations are missense mutations. Untreated PKU can lead to intellectual disability, seizures, and other symptoms. Various single nucleotide variations (SNVs) can cause PKU (27). SNVs may affect folding, expression, binding affinity, post-translational modification, and other protein features (28). We can use bioinformatics tools to gather medical and biological information in order to predict the pathogenicity of different genetic variations (29). Thusberg et al. evaluated the performance of nine pathogenicity prediction tools including Panther, MutPred, PhD‐SNP, PolyPhen, SNAP, PolyPhen2, SIFT, nsSNPAnalyzer, and SNPs&GO using different human SNP databases; they disclosed that SNPs&GO was the best in accuracy and SNAP was better in NPV. They totally found MutPred and SNPs&GO the most reliable prediction tools for variants. They believe that there is no single method as the best tool for all parameters, so selecting the proper software and tools depends on the nature of the data (30). In the present study, we used many bioinformatics software packages and online tools to evaluate the mutations and the protein structure in different aspects and obtain more reliable results by using an acceptable number of tools.
In our previous study, PAH mutations within exon 6, exon 7, and exons 10 - 12 were identified in 40 PKU patients in the southwest of Iran. The results showed one novel variation besides some previously reported ones. Q375R (c.1124A>G) was detected in exon 11 as a novel variant. Seven different polymorphisms in exonic regions and three new variants in intron regions of PAH were detected (14). In this study, we aimed to evaluate new variation potentiality for pathogenicity using various software packages and databases. The structural characteristics, stability, post-translational modifications (data not shown), and pathogenicity change predictions were checked for this new variation. The results showed that new variation Q375R is disease-causing according to the MutationTaster, polyphen2, and SIFT; however, other tools predicted this variation is neutral or intermediate damaging.
Much evidence implicates protein misfolding in human diseases. Indeed, normal, correctly folded proteins are transformed from their proper structure into an abnormal structure, amyloid fibril (20). Q375R mutation decreases the amyloid propensity and the stability of the protein, and changes the residues in the binding site. Chaperon binding to the protein in PAH could take place due to the effect of this variation. Therefore, the variation could affect the half-life, folding, and functionality of PAH. However, the mutation did not affect the aggregation tendency and post-translational modifications of the protein. Despite the functional assays necessity for variation evaluation, these predictions shed light on the path of these assessments.
Finally, it can be concluded that the new variation Q375R is likely deleterious and PKU causing; so, this variant could be used to diagnose PKU. In addition, the results showed that this residue alteration potentially affected some structural and stability features of PAH. However, in vitro and in vivo functional evaluation can provide certainty to this end and clarify some controversial and ambiguous results of predictive tools.