





1. Background
Hepatitis C virus (HCV) is a major cause of liver diseases. As a major cause of cirrhosis and liver carcinoma, chronic HCV infection occurs in 70-80% of people. It is estimated that the risk of cirrhosis and liver carcinoma ranges from 15% to 30%. Globally, 71 million people suffer from chronic HCV infection, with an annual mortality rate of 3% (1, 2). Transmission of HCV, as a blood-borne virus, is the major concern for the safety of blood products. In recent decades, several strategies, such as sensitive blood screening tests and donor selection, are developed to increase the safety of blood products (3). The prevalence of HCV among Iranian blood donors is approximately one-tenth of its prevalence in the general population (0.05% vs. 0.6%), mainly due to the efforts made by the Iranian Blood Transfusion Organization (IBTO) (3, 4). Contact with small quantities of infected blood is the main transmission route of HCV. A recent study on Iranian blood donors showed that intravenous drug use (IDU) is the main risk factor for HCV infection (5).
HCV is a family of Flaviviridae with a positive-sense single-stranded RNA of 9,600 bp in length. Due to the high rate of mutation in the NS5B region of HCV genome coding RNA-dependent RNA polymerase enzyme, its genetic heterogeneity can be explained. HCV genome is classified into 8 major genotypes and 90 Sub- genotypes (6). Epidemiologically, an association between HCV genotype and HCV transmission route has been reported. In Europe, the most frequent type of HCV among transfusion-related HCV infection patients is genotype 1b, while for IDUs, the genotype 3a is dominant (7, 8). In Iran, among patients with transfusion-related risk, inherited bleeding disorders (IBD) patients are reported to have a high rate of HCV infection. Sequence analysis of the NS5B region of HCV showed that genotype 1a is the most frequent subtype, followed by subtype 3a, in transfusion-related HCV infection patients (9, 10), and genotype 3a is reported to be the most common genotype in IDUs and blood donors (11-13).
Increasing awareness about HCV transmission is of crucial importance for the successful implementation of strategies intended to prevent the spread of HCV. For nearly four decades, molecular epidemiology has been used as an important tool for investigating the spread of infectious diseases (14). Phylogenetic analysis can be used to precisely investigate the spread of infectious agents (15). By phylogenetic analysis, the sequences are compared with each other to investigate the association between the sequences. Recently, changing the molecular epidemiology of HCV infection has been suggested. Sequence analysis of NS5B region of HCV among Iranian blood donors showed replacement of genotype 1a by genotype 3a (13).
2. Objectives
To investigate the association between HCV genotype 3a circulating in the community and obtaining a clear perspective on changing the molecular epidemiology of HCV among Iranian blood donors, in the presented study, a partial region of NS5B sequences derived from Iranian blood donors and two high-risk populations (i.e., IDUs and IBDs) have been compared.
3. Methods
3.1. Study Population
In total, from 2016 to 2017, 110 HCV sequences were obtained from Iranian blood donors from all corners of the country and were submitted to GenBank by the authors in 2017 (13). The sequences were corresponding to nucleotides 8256 - 8636 of the NS5B region of the HCV genome (A 380 bp segment). Fifty-eight sequences that contained HCV genotype 3a were included in the study. In this cross-sectional study, all 58 HCV sequences with the accession number of MG704694 to MG704794 were compared with the same NS5B partial sequences of HCV genotype 3a obtained from IDUs and IBDs. The later sequences had been submitted to GenBank with the accession number of JQ433655- JQ433608 and KC351605- KC351634, respectively, and their manuscripts were published in 2012 and 2016 (10, 11, 13). Based on accession numbers available on the related manuscript, the FASTA formats of NS5B sequences of IDUs and IBDs were extracted from the GenBank database. Data on the history of intravenous injection in blood donors were obtained from a post-donation questionnaire (5).
3.2. Phylogenetic Analyses
First, sequences of 58 blood donors were aligned to 48 Iranian IDU sequences using the ClustalW program, and a phylogenetic tree was constructed. Second, sequences of 58 blood donors, 48 IDUs, and 31 IBDs were aligned, and another phylogenetic tree was formed. Phylogenetic trees were constructed using MEGA X (16) by UPGMA method based on Kimura 2-parameter distances method with bootstrap tests of 1000 replications (17, 18).
3.3. Ethics Approval and Consent to Participate
The current study was approved by the Ethics Committee of High Institute for Research and Education in Transfusion Medicine, Tehran, Iran (code No: IR.TMI.REC.1394.1800). Written informed consent was obtained from all participants.
4. Results
Of 58 blood donors with subtype 3a, 31 (53.45%) were reported risk of intravenous injection (BDs/IDUs), whereas no history of IDU was reported by other subjects (BDs). Two blood donors had identical sequences (donor 67 and donor 68) and were from the same city.
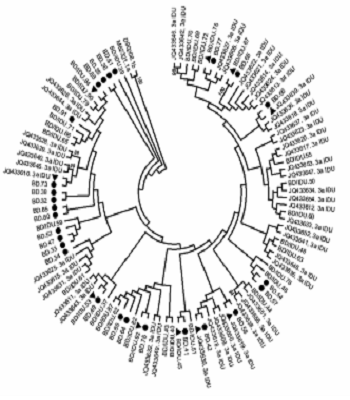
A phylogenetic tree was constructed with HCV strains with genotype 3a from all donors and IDUs (Figure 1). No separate cluster was formed to differentiate HCV sequences in BDs, BDs/IDUs, and IDUs. Sequences of BDs were phylogenetically intermixed with IDU sequences (BD/IDU and IDU sequences). Of 27 strains from donors who did not report intravenous injection (BDs), 20 sequences formed 15 small clusters, including five clusters (33%) with BD sequences and 10 clusters (67%) with sequences of BDs/IDU or IDUs. The remained sequences generated from BDs were indistinctly related to other strains.
, and blood donors who reported IDU risk (BD/IDU) and 48 injecting drug users (IDU) retrieved from GenBank. Accession numbers of IDU sequences were shown. In the small clusters, sequences of BDs were indicated by symbols of ● and sequences of BD/IDU, and IDUs were indicated ▲ in the small clusters. Bootstrap values based on 1000 replicated > 70 are given at the branches.")
phylogenetic tree of HCV subtype 3a strains was constructed by UPMEGA kimura-2 parameter model based on partial sequences of NS5B region of HCV genome from 58 blood donors including of blood donors denied to report IDU risk (BD), and blood donors who reported IDU risk (BD/IDU) and 48 injecting drug users (IDU) retrieved from GenBank. Accession numbers of IDU sequences were shown. In the small clusters, sequences of BDs were indicated by symbols of ● and sequences of BD/IDU, and IDUs were indicated ▲ in the small clusters. Bootstrap values based on 1000 replicated > 70 are given at the branches.
Another phylogenetic tree was formed by HCV genotype 3a sequences of 58 blood donors, 48 IDUs, and 31 IBDs to find any association between sequences (Figure 2). For more clarity, the upper part of figure (A) is shown in Figure 2A, and the rest is shown in Figure 2B. A middle level of clustering was found in this tree. Thirty-eight strains (out of 58) generated from blood donors, including sequences of 19 BD, 19 BD/IDUs, and 14 IDUs, formed a cluster in the tree (Figure 2A). None of the IBD sequences were included in cluster A. In contrast, all 31 IBD strains were located in remained parts of the tree along with the majority of IDU strains (30/48), and most of the strains were isolated from blood donors, including 8 from BDs and 13 from BDs/IDUs. IBDs sequences formed 17 small clusters including 9 (52.94%) with BDs/IDUs and IDU sequences, 6 (35.29%) with IBD sequences, and 2 (11.76%) with BD sequences (Figure 2B). According to the results, IBD sequences were more close to sequences of BDs/IDUs and IDU than those isolated from BDs (88.64% compared to 11.76%).
 and blood donors who reported IDU risk (BD/IDU), 48 injecting drug users (IDU) and 31 inherited bleeding disorders patients (IBD) retrieved from GenBank. Accession numbers of IDU and IBD sequences were shown. In the small clusters BD, BD/IDU, IDU, and IBD sequences were indicated by symbols of ●, ▲, ■ and ▼. Bootstrap values based on 1000 replicated > 70 are given at the branches. The tree was illustrated in 2a for the upper part and 2b for the rest as described in the text.")
phylogenetic tree of HCV subtype 3a strains was constructed by UPMEGA kimura-2 parameter model based on partial sequences of NS5B region of HCV genome from 58 blood donors including of blood donors denied to report IDU risk (BD) and blood donors who reported IDU risk (BD/IDU), 48 injecting drug users (IDU) and 31 inherited bleeding disorders patients (IBD) retrieved from GenBank. Accession numbers of IDU and IBD sequences were shown. In the small clusters BD, BD/IDU, IDU, and IBD sequences were indicated by symbols of ●, ▲, ■ and ▼. Bootstrap values based on 1000 replicated > 70 are given at the branches. The tree was illustrated in 2a for the upper part and 2b for the rest as described in the text.
5. Discussion
Some recently conducted studies reported changes in the distribution of HCV genotype as replacement of genotype 3a, instead of genotype 1a, as the most common genotype among Iranian blood donors (13). In the presented study, the phylogenetic tree (Figure 1) revealed that genotype 3a sequences isolated from blood donors were not genetically different from that of IDUs, which is similar to the study conducted on blood donors in the Netherland (19). The reported phylogenetic level of 3a sequences between blood donors and IDUs supported the occasional transmission of IDU-related strain 3a into the general population and suggested the circulation of HCV with genotype 3a in the general population through intravenous drug injection or other modes of transmission. By excluding the possibility of not reporting the risk of intravenous injection by BDs, it seems that BDs may be infected through high-risk behaviors or parenteral iatrogenic procedures. A study in China noted the role of IDUs in altering the molecular epidemiology of HCV in HIV/HCV co-infection (20).
As before IBTO establishment, blood was sold to patients, and IDUs were the largest blood sellers, it is possible that IDU-related genotype 3a has transmitted to the general population through donating infected of blood IDUS and caused the explosion of genotype 3a, as it has already suggested (10). This phenomenon has gradually resulted in the similarity of predominant HCV genotype among IDUs and blood donors.
Another phylogenetic tree showed the phylogenetic mixing of genotype 3a sequences among blood donors, IDUs, and IBDs representing the spreading of genotype 3a sequences as an IDU-related genotype among the Iranian population. Furthermore, a small cluster made by similar sequences from two blood donors (donor 67 and donor 68) provided strong evidence of local HCV transmission among blood donors from the same geographical region.
As a result of implementing various preventive methods such as screening donors and analyzing blood samples for various agents, including HCV, the risk of HCV transmission declined in recent decades (3, 4). According to the IBTO policy, potential blood donors with a history of intravenous injection are permanently deferred from donation. Nevertheless, these programs have not been successful always, so that some IDUs have passed these programs and donated their blood. Such donors can increase the risk of HCV transmission among recipients. Since, currently, no vaccine is avaiable for HCV infection and HCV genotype 3 is the most difficult genotype in treatment even with new direct-acting antivirals (DAA) as well as the high rate of HCV infection progression and mortality rate related to the genotype 3 (21, 22), prevention of HCV transmission, especially HCV genotype 3a, via blood transfusion should be considered as a priority to improve the safety of blood products. Comparing the sequences of the NS5B region of HCV in Iranian blood donors and the corresponded sequences derived from IDUs and IBD patients, which are submitted to the GenBank and published as scientific articles, may cause bias in phylogenetic analysis. In the present study, comparing the donor sequences of the NS5B gen related to HCV genotype 3a with the corresponding sequences of IDUs and IBDs obtained from the published study has probably caused biases in the phylogenetic analysis results.
5.1. Conclusion
The phylogenetic tree showed the phylogenetic mixing of genotype 3a sequences in blood donors, IDUs, and IBD patients. The results confirmed the extensive transmission of IDU-related genotype 3a into the general population.
According to the IBTO policy, potential blood donors with a history of intravenous injection are permanently deferred from donation. Nevertheless, these programs have not been successful always, so that some IDUs have passed these programs and donated their blood. Such donors can increase the risk of transmission of HCV genotype 3a in the community. More attention should be paid to the blood donor selection.
