




1. Background
2. Objectives
3. Methods
3.1. Cell Extraction
3.2. Cell Culture
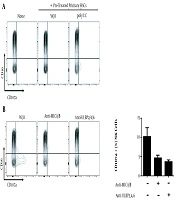
 Primary HSCs were pretreated with Anti-MICA/B and Anti-ULBP2,4,6 before poly I:C stimulation, then cocultured with NK cells, CD107a expression was shown. w/o means without anti-MICA/B or anti-ULBP2/4/6 (B).")
Senescent HSCs trigger immunosurveillance from NK cells through upregulated NKG2D ligands. Primary HSCs stimulated with poly I:C were cocultured with NK cells, the expression of CD107a was detected by flow cytometry. None means only NK cells, w/o means without poly I:C. (A) Primary HSCs were pretreated with Anti-MICA/B and Anti-ULBP2,4,6 before poly I:C stimulation, then cocultured with NK cells, CD107a expression was shown. w/o means without anti-MICA/B or anti-ULBP2/4/6 (B).
3.3. Flow Cytometry
3.4. Immunoblotting
3.5. Real-Time PCR
3.6. SA-β-gal Staining
3.7. Statistical Analysis
4. Results
4.1. TLR3 Mediates Senescence in HSCs

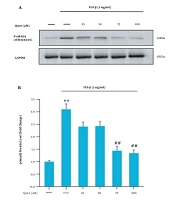
TLR3 activation induces cellular senescence in HSCs. HSCs were treated with poly I:C at indicated concentration. A, TLR3 expression was measured by qRT-PCR; B, p16 and p21 expression was measured by qRT-PCR; C, Western blot analyses of protein expression of senescence molecules P16, P21 and fibrogenic molecule α-SMA; D, Senescence-dependent β-galactosidase activity staining showing significant upregulation in the poly I:C-treated HSCs. **, P < 0.01; ***, P < 0.001.
4.2. Increased NKG2D Ligand Expression in TLR3-Induced Senescent HSCs

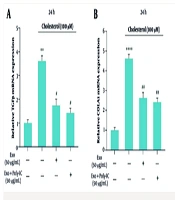
NKG2D ligands expression was increased in TLR3-induced senescent HSCs. HSCs were treated with poly I:C at indicated concentration. A, MICA and ULBP expression in LX2 cells were measured by qRT-PCR; B, The expression of MICA/B, ULBP and DcR2 in LX2 cells was analyzed by flow cytometry; C, MICA, MICB and ULBP expression in primary HSCs was measured by qRT-PCR. **, P < 0.01; ***, P < 0.001. ns, not significant.