










1. Background
Limited sources of fresh water necessitate the application of health policies for treatment and decontamination of human sewage in order to be used as an alternative for several purposes, including land application, irrigation of green spaces, fire systems, and industrial use (1). As the largest biological sink, sewage contains a pool of infectious agents as well as other organic and non-organic materials. A wide variety of infectious agents, including bacteria, fungi, parasites, and viruses can be found in sewage, which makes it an important transmission vehicle (2). Enteric viruses, including some members of rotaviruses, enteroviruses, astroviruses, noroviruses, adenoviruses, hepatitis A virus (HAV), and hepatitis E virus, are transmitted mainly through the fecal-oral route (3). Non-enveloped nature of these viruses provides them with the potential to resist different environmental pressures and keeps them in an active state (4). Considering the inability of the current decontamination strategies to clear viruses from treated effluent, viruses can contaminate drinking water sources and transmit to humans and cause disease in them. For instance, Adegoke et al. reported the transmission of viruses via contaminated effluent-irrigated crops and subsequent large epidemics (5).
HAV belongs to Picornaviridae family of the order Picornavirales. Viruses of this family contain a single-stranded positive sense linear RNA with a size of 7.5 kb and a single open reading frame (ORF), which encodes structural and non-structural proteins required for virus replication (6, 7). The genome is encapsidated in an icosahedral protein shell of approximately 27 nm in diameter, consisting of viral protein 1 (VP1), VP2, VP3, and VP4 (8).
The virus is classified in the genus Hepatovirus. Based on the genetic variability, this genus is divided into six genotypes (9). Genotypes I, II, and III have been isolated from human, and the other genotypes have been detected in simians (10). Subgenotypes have also been specified for further classification. Genotype 1 is the most prevalent genotype all around the world. Moreover, the prevalence of subgenotype IA predominates IB (11). Global distribution of different genotypes and subgenotypes varies in different regions (12-14). The most reported subgenotype in South and North America, Europe, Asia, and Africa is subgenotype IA, and in Australia, the Middle East, and South Africa, subgenotype IB predominates. Albeit, co-circulation of multiple genotypes has also been documented in different geographical regions (15, 16).
The current strategies for treatment of human sewage appear to be inefficient in removing viral particles. Moreover, growing evidence about the persistence of pathogenic viruses in water environments necessitates re-evaluation and revision of the current protocols on the assessment of the wastewater suitability for further use (5, 17).
2. Objectives
This study aimed to evaluate HAV contamination in human sewage before and after treatment in all different seasons of 2019 in the wastewater treatment plant of Ekbatan town in Tehran, Iran, and analyze the phylogenetic properties of the identified viruses.
3. Methods
3.1. Sample Collection
In this study, 50 mL of wastewater samples were collected between 7:00 to 9:00 a.m. in sterile tubes. A total of 18 wastewater samples were collected from the wastewater treatment plant of Ekbatan town in Tehran, Iran, in 2019. This plant is operated by a secondary biological treatment with activated sludge coupled with chlorine disinfection, and it is expected to cover the wastewater generated by 100,000 residents in western regions of Tehran. Samples were collected from three sites, including entry points (as influent), before chlorination, and exit points (as effluent), and were transferred to the laboratory in cold boxes. All samples were collected in duplicates and filtered as soon as they were delivered to the laboratory.
3.2. Extraction of Viral RNA
Viral RNA was extracted directly from 200 µL of wastewater samples, using a FavorPrep™ Total RNA Mini Kit (Favorgen, Ping-Tung, Taiwan) following the manufacturer’s instructions. Briefly, 200 µL of samples were applied directly to a filter column and centrifuged at full speed (~ 18000 x g) for 2 min. One volume of 70% ethanol was added to the flow-through and mixed by vortexing. The sample mixture was transferred to the mini column and centrifuged at full speed for 1 min. After three washing steps, the column was centrifuged at full speed for an additional 3 min to dry the column. The RNA was eluted in 50 µL of RNase-free H2O and stored at -70°C for further analysis. It should be noted that the extraction was performed directly from raw sewage samples without virus concentration.
3.3. Complementary DNA Synthesis
Complementary DNA was synthesized using a complementary deoxyribonucleic acid (cDNA) synthesis kit (Favorgen, Ping-Tung, Taiwan). For this aim, 5 µL of the extracted RNA was used as a template. Then, 1 µL random hexamer and 7.4 µL of DEPC-treated water were added to the extracted RNA. The mixture was incubated at 70°C for 5 min. Next, 4 µL of 5X first-strand buffer, 1 µL of dNTPs (10 mM each), 0.5 µL of RNasin (40U/µL), and 1 µL of Moloney murine leukemia virus (M-MLV) reverse transcriptase were added to a 20-µL reaction mixture. All steps were performed on ice. The mixture was then incubated for 60 min at 37°C, and the reaction was terminated by heating at 70°C for 5 min. The product was stored at -20°C until use.
3.4. Polymerase Chain Reaction (PCR)
Semi-nested PCR test was used to identify HAV genome in wastewater samples. Specific primers were employed for the amplification of a 222-bp fragment in VP3/VP1 junction of the viral genome (18). The forward and reverse primers for the first round PCR were 5´-CAGGAAATGTCTCAGGTACTTTCT-3´ and 5´-GCTCCTCTTTATCATGCTATGGAT-3´, respectively. The amplification conditions of this step included an initial denaturation at 94 °C for 5 min, 40 cycles at 94°C for 45 s, 57°C for 1 min, and 72°C for 1 min, and a final elongation step at 72°C for 10 min. The forward and reverse primers for the semi-nested step were 5´-CAGGAAATGTCTCAGGTACTTTCT-3´ and 5´-ATGTTACTACACAAGTTGGAGAT-3´, respectively. The amplification conditions for this step included an initial denaturation at 94°C for 5 min, 40 cycles at 94°C for 45 s, 55°C for 1 min, and 72°C for 1 min, and a final elongation step at 72°C for 10 min. The amplified fragments were resolved on a 1% agarose gel and visualized under ultraviolet (UV) light using a Gel-Doc apparatus (Vilber Lourmat, Gel Imaging system E Box CX5, France). A 100-bp DNA size marker was used as the reference for 222-bp amplicon. A commercially synthesized VP3/VP1 junction gene cloned in pUC57 plasmid (Gene Universal Inc. Newark, DE, USA) was used as a positive control. No-template and nuclease-free water were used as negative controls.
3.5. Phylogenetic Analysis
The amplified PCR products were purified using a QIAquick PCR purification kit (Qiagen, Chatsworth, CA) and sequenced using Big-Dye terminator sequencing and an ABI 3730XL DNA analyzer (Applied Biosystems, Foster City CA, USA).
The obtained sequences were evaluated by Basic Local Alignment Search Tool (BLAST), analyzed, and trimmed using MEGA 10 software, and multiple sequence alignment was performed using Clustal W algorithm with the reference sequences retrieved from GenBank.
For phylogenetic analysis, the sequences obtained in this study plus the following 25 reference sequences were analyzed: sub-genotype IA (GenBank: EF406357; KC182588; X75215; AB623053), sub-genotype IB (GenBank:AF314208; M20273; NC001489; AF268396; MK829707; LC128713; HQ246217; KX228694; JN388677; MG546668), sub-genotype IIA (GenBank: AY644676), sub-genotype IIB (GenBank: AY644670), sub-genotype IIIA (GenBank: AJ299464; KC669704; DQ991029; AB279732), sub-genotype IIIB (GenBank: AB258387; AB300205; AB425339), and simian genotype V (GenBank: EU140838; D00924).
Phylogenetic analysis was performed using the MEGA10 software. Phylogenetic tree was constructed using the maximum likelihood (ML) algorithm and bootstrap resampling with 1000 replicates. Nucleotide substitutions and substitution rates were calculated using the General Time Reversible + G + I (GTR-GI) model.
3.6. Nucleotide Sequence Accession Numbers
Nucleotides isolated from samples in this study were stored in the GenBank under the accession numbers MN648638-MN648647.
4. Results
4.1. Virus Detection in Sewage Samples
HAV was detected in 15/18 (~ 83%) of sewage samples (Table 1). All influent samples were positive for the virus throughout the year. All the samples before chlorination were also positive, while in 3/6 (50%) of effluent samples the virus was not identified. All the negative samples had been taken from the treatment plant during spring and winter.
| Variables | Sampling Seasons and Dates | |||||
|---|---|---|---|---|---|---|
| Spring | Summer | Autumn | Winter | |||
| 5 May | 21 Jul. | 6 Sep. | 19 Dec. | 7 Jan. | 6 Mar. | |
| Influent | + | + | + | + | + | + |
| Before chlorination | + | + | + | + | + | + |
| Effluent | - | + | + | + | - | - |
4.2. Phylogenetic Analysis of HAV
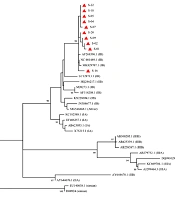
Among the 15 HAV-positive samples, 10 were subjected to sequencing. Phylogenetic analysis of a 222-bp region in VP3/VP1 junction of the genome was performed. Based on the results of this analysis, all the identified genotypes were classified as subgenotype IB group (Figure 1).
 in influent and effluent samples of the wastewater treatment plant of Ekbatan town in Tehran. The tree was constructed using ML algorithm and GTR-GI model (MEGA10) with a bootstrap of 1000 replicates. Bar, 0.05 substitutions per nucleotide position")
Phylogenetic analysis of the detected HAV genotypes (red rectangles) in influent and effluent samples of the wastewater treatment plant of Ekbatan town in Tehran. The tree was constructed using ML algorithm and GTR-GI model (MEGA10) with a bootstrap of 1000 replicates. Bar, 0.05 substitutions per nucleotide position
5. Discussion
After treatment based on the current guidelines, wastewater is used for different purposes with a final discharge to the environment (1). Since the application of any protocol to eliminate viral contaminations has not been mandatory so far, watstewater has always been considered as a possible source of outbreaks of human viral diseases (19, 20). Besides, since most of the enteric viruses are shed in high concentrations in the feces and have a low infectious dose, contaminated water sources can play a major role in transmission of different pathogenic viruses to a susceptible human host, especially in regions with poor sanitary infrastructures (21, 22).
HAV is responsible for the majority of water-borne and food-borne outbreaks of hepatitis (23). Like other enteric viruses, high titers of the virus are shed in the feces of the affected person. These particles not only can survive routine procedures of wastewater treatment, but also they are resistant to different environmental conditions and retain their infectivity in soil, food, shellfish, and water sources for a long time (24, 25). Transmission through fecal-oral route and low infectious dose enables the virus to infect new susceptible hosts and start a new infection cycle (16). Therefore, investigating the presence of HAV in sewage provides a general picture of the virus spread in an entire population. Moreover, this can be a representative of symptomatic and asymptomatic infections in a particular region revealing the circulation of HAV between the environment and the human population.
In the present study, RT-PCR was conducted to investigate the presence of HAV in sewage samples of a wastewater treatment in Tehran. During a one-year surveillance, HAV was detected in all influent samples, which indicates that the infection is endemic in this area all year round. Since no approach for viral particle concentration was applied to the samples, these results may reflect the high viral loads in the samples. The virus was also detected in all treated samples before chlorination, which suggests that pre-chlorination processes are unable to eliminate the virus from sewage. However, HAV was not detected in 50% of effluent samples. This result might be due to the adverse effect of the chlorine on virus particles. On the other hand, HAV detection in 50% of the effluent samples indicates that chlorine concentration (0.5 to 1.5 ppm) might not be sufficient to inactivate all the particles (24, 26). Besides, the detection of the virus during autumn and summer, and not in winter and spring, suggests that the titers of HAV may have increased during those two seasons. Therefore, the level of chlorine might have not been enough to degrade all the particles. However, it should be mentioned that if virus concentration by filtration was performed on the samples, virus titers might be increased to detectable levels of PCR assay, at least in some of the effluent samples.
Some evidence suggests that current sewage treatment methods, such as solar radiation or chlorination, are able to inactivate virions without degrading virus structure or genome integrity (27). Therefore, it should be noted that the detection of viral genome through molecular tests does not necessarily represent the infectious nature of the detected particles in environment and may overestimate the actual attributed threat to the human health.
One of the restrictions of this study was that the gold standard to assess the infectivity of detected viruses is cell culture; however, enteric viruses, including HAV, are difficult to culture (28). Moreover, virus quantification via real-time qPCR assay may help to better evaluate the effect of different processes of sewage treatment in the reduction of viral load.
Seroprevalence data in Iran shows high HAV endemicity with levels ranging between 8 and 99% (29), which confirms that Iran is an endemic region for HAV infection. Phylogenetic analyses of HAV sequences (VP3/VP1 junction) in this study showed the predominance of subgenotype IB, which is in accordance with the results of Nejati et al. (30) in Ahvaz, Iran. This is also in accordance with the general pattern of HAV genotype distribution in the Middle East with the predominance of subgenotype IB (15).
5.1. Conclusions
The results of this study indicated the presence of high titers of HAV in sewage and the inability of the current wastewater treatment protocols in removing the viral particles, and a need for revision of these protocols. These findings may indirectly reflect the high endemicity of HAV infection of subgenotype IB in Ekbatan town of Tehran throughout the year. Based on the results of this study, to restrict viral spread in the environment, it is highly suggested that the present guidelines be reassessed.
