




1. Background
Hepatitis B virus (HBV) as a hepatotropic virus may result in severe liver diseases, such as hepatitis, cirrhosis, and hepatocellular carcinoma. Currently, HBV infection is still one of the global public health problems (1). Despite the availability of vaccines, approximately 3.5% of the population in the world is chronically infected with HBV (1). Patients with chronic HBV infection are treated with nucleoside analogs and/or interferon-alpha in a clinic. However, this medication can only inhibit HBV replication but cannot eradicate it (2). The microenvironment of the liver may be connected with persistent HBV infection. However, the mechanisms by which HBV initiates chronic infection are not fully defined yet (3).
Hepatitis B virus is an incompletely double-stranded hepatotropic virus whose genome contains four overlapping reading frames (4). After entry into the hepatocytes, the HBV genome is repaired by host enzymes needed to synthesize a covalently closed circular genome (cccDNA), the template for viral mRNA transcription. The HBV genome encodes hepatitis B core antigen (HBcAg), viral envelope proteins known as surface antigens (HBsAg), regulatory protein X, hepatitis B e antigen (HBeAg), and viral polymerase which is involved in DNA synthesis and RNA encapsidation (4). All these structural or non-structural proteins may modulate immune response or induce immune tolerance (5). Hepatitis B e antigen, an essential regulator of innate and/or adaptive immunological responses, is secreted from the liver into the circulatory system during HBV infection, which is responsible for maintaining persistent infection (6).
As pattern recognition receptors, toll-like receptors (TLRs) are essential for identifying evolutionarily conserved pathogen-associated molecular patterns (PAMP). They play a vital role in inherent immunity (7). Sensing and activation of TLR result in innate antiviral responses in vivo (8). Visvanathan and colleagues demonstrated that the expression and function of TLR2 in the livers of HBeAg-positive patients was decreased, and this down-regulation was concomitant with an increase of HBeAg (9). However, it is unknown which factor is operating in this condition (10).
Bile acids are essential physiological agents for lipid absorption and perform the function of signaling molecules that maintain metabolic homeostasis (11). They are synthesized from cholesterol in the liver and then excreted into the bile. In the presence of intestinal microbiota, primary bile acids are converted into secondary bile acids. In the enterocytes from the distal ileum, bile acids are then avidly re-absorbed. Bile acids in the portal blood are absorbed by the hepatocytes and re-secreted to the lumen. The enterohepatic circulation played a considerable part in balancing bile acid synthesis, secretion, and re-absorption (12). Recent studies have revealed that bile acids are potent signaling molecules that regulate metabolism, immune response, and inflammation, predominantly mainly through nuclear receptor farnesoid X receptor (FXR) and G protein-coupled receptor (TGR5) (13).
It has been known that the concentration of serum bile acids is remarkably elevated during acute or chronic HBV infection (14), which is often regarded as a marker of liver damage, just like aminotransferase. Interestingly, it is reported that HBV infection can noticeably induce the expression of cholesterol 7a-hydroxylase (CYP7A1) through the binding of HBV to Na+-taurocholate co-transporting polypeptide (NTCP) in HBV-infected mice (15). However, the molecular mechanisms of virus-host interactions during chronic HBV infection remain largely unexplored (16).
2. Objectives
In the current study, we aimed to probe whether and how the bile acids modulate HBV expression and relative immune responses.
3. Methods
3.1. Mice and Treatment
Male C57BL/6 mice, 6 - 8 weeks old, were purchased from Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). FXR-/- mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All mice were fed a standard chow diet and maintained in specific pathogen-free (SPF) conditions. All animal studies were approved by the Institutional Animal Care and Use Committee at Henan University (Kaifeng, China). This study was completed in strict accordance with the guidelines of the Guide for the Care and Use of Laboratory Animals, in compliance with the regulations of the People’s Republic of China. Hydrodynamic injections were conducted as described above (17). We injected 10 μg of pAAV/HBV1.2 plasmid in a volume of 0.9% NaCl into the tail vein of mice up to 10% of mouse body weight within 5 - 8 seconds. Then, we collected serum specimens and liver tissueat the specified time points.
3.2. Plasmids and Reagents
The plasmids of pAAV/HBV1.2, which encode all HBV messages and mutant HBeAg-null pAAV/HBV1.2, were kindly provided by Prof. Pei-Jer Chen (National Taiwan University, Taiwan). Plasmid pHY106+wta containing full-length HBV genome was provided by Prof. Zhongji Meng (Hubei University of Medicine, Shiyan, China). The plasmids of pGL3-EN2/Cp (HBV enhancer II/core promoter-reporter), phFXR (containing the full-length human FXR gene), and pRL-TK were constructed and stored in our laboratory. Farnesoid X receptor agonists chenodeoxycholic acid (CDCA) and GW4064 and FXR antagonist Z-guggulsterone (Z-g) were purchased from Sigma (St. Louis, MO).
3.3. Cell Culture and Transfection
The human hepatoma HepG2 cell line and human embryonic kidney HEK293T cell line were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum at 37°C in 5% CO2. The cells were transfected with plasmids at indicated concentrations using lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. At 24 h after transfection, the cells were treated with CDCA, GW4064, or Z-g at indicated concentrations for 24 or 48 h.
3.4. RNA Isolation and Reverse-transcription-Quantitative PCR
Total RNA was extracted from HepG2 cells or liver tissues using TRIzol reagent (Invitrogen). Reverse-transcription (RT)-quantitative (q) PCR analysis was performed using the ABI 7300 Sequence Detection System. Results were calculated using Ct values and normalized to the 36B4 mRNA level. All primer sequences are listed in Table 1.
| Gene | Primer Sequences |
|---|---|
| TLR2 (mouse) | |
| Forward | 5’-GCTCCTGCGAACTCCTATCC-3’ |
| Reverse | 5’-CAGCAGACTCCAGACACCAG-3’ |
| TLR3 (mouse) | |
| Forward | 5’-GTGAGATACAACGTAGCTGACTG-3’ |
| Reverse | 5’-TCCTGCATCCAAGATAGCAAGT-3’ |
| TLR4 (mouse) | |
| Forward | 5’-ATGGCATGGCTTACACCACC-3’ |
| Reverse | 5’-GAGGCCAATTTTGTCTCCACA-3’ |
| TLR9 (mouse) | |
| Forward | 5’-ATGGTTCTCCGTCGAAGGACT-3’ |
| Reverse | 5’-GAGGCTTCAGCTCACAGGG-3’ |
| 36B4 (mouse) | |
| Forward | 5’-AGATTCGGGATATGCTGTTGGC-3’ |
| Reverse | 5’-TCGGGTCCTAGACCAGTGTTC-3’ |
| TLR2 (human) | |
| Forward | 5’-GCGTTCTCTCAGGTGACTGCTCG-3’ |
| Reverse | 5’-GAAAGCAGTGAAAGAGCAATGGGCACAA-3’ |
| TLR3 (human) | |
| Forward | 5’-ACAACTTAGCACGGCTCTGGA-3’ |
| Reverse | 5’-ACCTCAACTGGGATCTCGTCA-3’ |
| TLR4 (human) | |
| Forward | 5’-AATCTAGAGCACTTGGACCTTTCC-3’ |
| Reverse | 5’-GGGTTCAGGGACAGGTCTAAAGA-3’ |
| TLR9 (human) | |
| Forward | 5’-GGACCTCTGGTACTGCTTCCA-3’ |
| Reverse | 5’-AAGCTCGTTGTACACCCAGTCT-3’ |
| β-actin (human) | |
| Forward | 5’-TTGTTACAGGAAGTCCCTTGCC-3’ |
| Reverse | 5’-ATGCTATCACCTCCCCTGTGTG-3’ |
Reverse-transcription-PCR Primer Sequences
3.5. Enzyme-linked Immunosorbent Assay for HBeAg Quantitation
The amount secreted in the supernatants and intracellular HBeAg was determined using the commercially available enzyme-linked immunosorbent assay (ELISA) kit (Kehua Bio-engineering Co., Shanghai, China) according to the manufacturer’s instructions. Mouse serum samples were with a dilution of 1: 10 and then were used for HBeAg detection.
3.6. Dual-luciferase Reporter Assay
For luciferase assay, HepG2 cells were transiently transfected with plasmids pGL3-EN2/Cp, pRL-TK, and/or phFXR. At 24 h after transfection, the cells were treated with 10 μM CDCA or vehicle (DMSO) for 48 h. Finally, the treated cells were harvested and lysed, followed by determining the luciferase activity using a dual-luciferase reporter assay system following the manufacturer’s instructions (Promega, Madison, WI). Luciferase activities were normalized by cotransfection with pRL-TK.
3.7. Statistical Analysis
The data from at least three independent experiments are expressed as the mean ± SD. The data were analyzed using the Student’s t-test. P < 0.05 was considered statistically significant.
4. Results
4.1. FXR Activation Promotes HBeAg Expression in Human Hepatoma HepG2 Cells
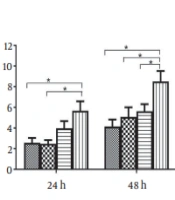
It has been shown that nuclear hormone receptors support HBV pregenomic (pg)-RNA synthesis and viral replication. However, how activation of FXR affects HBeAg expression and secretion is still unknown. Therefore, HepG2 cells were transiently transfected with plasmid pAAV/HBV1.2 and pGFP or phFXR. At 24 h post-transfection, cells were treated with the FXR agonist CDCA for 24 or 48 h. The results showed that FXR overexpression coupled with CDCA treatment elevated intracellular (Figure 1A) and extracellular (Figure 1B) levels of HBeAg in HepG2 cells. It suggests that the hepatoma cells which express FXR in nature can be activated in the presence of exogenous ligands. Moreover, we used another synthetic FXR agonist, GW4064, to confirm the effect of FXR activation on HBeAg expression. At 24 h after transfection, HepG2 cells were treated with GW4064 for 24 h. FXR overexpression plus GW4064 treatment also significantly upregulated intracellular (Figure 1C) and secreted (Figure 1D) HBeAg expression in HepG2 cells.
 agonist chenodeoxycholic acid (CDCA) and GW4064 promotes the expression and secretion of hepatitis B e antigen (HBeAg) in HepG2 and 293T cells. HepG2 cells in 24-well plates were transfected with pAAVHBV1.2 and/or phFXR and treated with CDCA (10 μM) or GW4064 (1 μM). Intracellular and extracellular HBeAg was measured with enzyme-linked immunosorbent assay (ELISA). A, The expression of intracellular HBeAg was detected after treatment with CDCA in HepG2 for 24 h and 48 h; B, Extracellular HBeAg after treatment with CDCA for 24 h and 48 h; C, Intracellular HBeAg after treatment with GW4064 for 24 h; D, Extracellular HBeAg after treatment with GW4064 for 24 h; E and F, Nonhepatoma cell line 293T was used as the hepatitis B virus (HBV) biosynthesis model; E, Intracellular HBeAg after treatment with CDCA for 48 h; F, Extracellular HBeAg after treatment with CDCA for 48 h. Three independent experiments were performed. * P < 0.05.")
Treatment with the farnesoid X receptor (FXR) agonist chenodeoxycholic acid (CDCA) and GW4064 promotes the expression and secretion of hepatitis B e antigen (HBeAg) in HepG2 and 293T cells. HepG2 cells in 24-well plates were transfected with pAAVHBV1.2 and/or phFXR and treated with CDCA (10 μM) or GW4064 (1 μM). Intracellular and extracellular HBeAg was measured with enzyme-linked immunosorbent assay (ELISA). A, The expression of intracellular HBeAg was detected after treatment with CDCA in HepG2 for 24 h and 48 h; B, Extracellular HBeAg after treatment with CDCA for 24 h and 48 h; C, Intracellular HBeAg after treatment with GW4064 for 24 h; D, Extracellular HBeAg after treatment with GW4064 for 24 h; E and F, Nonhepatoma cell line 293T was used as the hepatitis B virus (HBV) biosynthesis model; E, Intracellular HBeAg after treatment with CDCA for 48 h; F, Extracellular HBeAg after treatment with CDCA for 48 h. Three independent experiments were performed. * P < 0.05.
Considering that the plasmid backbone and HBV sequence may affect the expression of HBeAg, HepG2 cells were transfected with plasmid pHY106+wta, with or without CDCA treatment. Similar to the effects in the pAAV/HBV1.2 transfected cell model, CDCA also promoted intracellular HBeAg (Appendix 1) in pHY106+wta transfected cell systems. Collectively, FXR activation promotes HBeAg expression in HepG2 cells.
4.2. FXR Activation Promotes HBeAg Expression in Human Embryonic Kidney 293T Cells
In natural infections, HBV replication is principally limited to the liver, implying that specific liver-rich transcription factors control the propensity for HBV. We intended to investigate whether CDCA promotes HBeAg expression in non-liver cell lines. Compared to the control, CDCA failed to promote intracellular (Figure 1E) and secreted (Figure 1F) HBeAg expression in HEK293T cells. However, FXR overexpression coupled with CDCA treatment significantly increased HBeAg expression and secretion. Thus, there is ample evidence that FXR activation is sufficient to promote HBeAg expression in non-hepatoma cells.
4.3. FXR Inhibition Decreases the Promoting Effect of FXR Activation on HBeAg Expression
Z-guggulsterone is a natural product that reduces LDL cholesterol and is a potent antagonist of FXR (18). We wondered whether FXR inhibition decreases the promoting effect of FXR activation on HBeAg expression. In the presence of FXR agonist CDCA treatment, Z-g was decreased intracellular (Figure 2A) and secreted (Figure 2B) HBeAg expression in HepG2 cells. We also used HepG2 cells transfected with FXR siRNA to demonstrate that FXR knockdown had a similar reduction in HBeAg protein expression (data not shown). Z-guggulsterone did not modify the expression of HBeAg without FXR and CDCA (data not shown). Thus, FXR inhibition decreases the promoting effect of FXR activation on HBeAg expression.
, a natural farnesoid X receptor (FXR) α antagonist, inhibits the expression and secretion of hepatitis B e antigen (HBeAg) in HepG2. HepG2 cells transfected with pAAV/HBV1.2 and/or phFXR were incubated with chenodeoxycholic acid (CDCA) with or without Z-g for 48 h. After incubation, the concentration of HBeAg was detected by enzyme-linked immunosorbent assay (ELISA). A, Intracellular HBeAg; B, Extracellular HBeAg. Three independent experiments were performed. * P < 0.05.")
Z-guggulsterone (Z-g), a natural farnesoid X receptor (FXR) α antagonist, inhibits the expression and secretion of hepatitis B e antigen (HBeAg) in HepG2. HepG2 cells transfected with pAAV/HBV1.2 and/or phFXR were incubated with chenodeoxycholic acid (CDCA) with or without Z-g for 48 h. After incubation, the concentration of HBeAg was detected by enzyme-linked immunosorbent assay (ELISA). A, Intracellular HBeAg; B, Extracellular HBeAg. Three independent experiments were performed. * P < 0.05.
4.4. FXR Upregulates HBV Enhancer II and Core Promoter Activity
HepG2 cells were cotransfected with expression plasmids of pGL3-EN2/Cp, phRL-TK, and/or phFXR. Chenodeoxycholic acid at a concentration of 10 μM was added after 24 hours of transfection. In the absence of exogenous FXR, CDCA slightly increased the level of luciferase activity. In the presence of exogenous FXR, CDCA increased luciferase activity by approximately 450%, suggesting that FXR greatly influences the expression of HBeAg (Appendix 2). To further demonstrate if CDCA promotes HBV transcription, total RNA was extracted two days after adding CDCA and subjected to RT-PCR to analyze the synthesis of viral RNA. We designed a primer to distinguish precore and pregenomic RNA (preC/pgRNA) from other transcripts of HBV but could not distinguish between the two transcripts because of the overlap of precore and pregenomic RNA. Although exogenous expression of FXR or CDCA alone slightly promoted the synthesis of the preC/pgRNA, a notable increase in exogenous expression of FXR was observed with the lack of CDCA (Appendix 3).
4.5. Upregulation of HBeAg Renders Aberrant Toll-like Receptor 2 Expression by FXR
Toll-like receptors represent a type of pattern recognition receptors and control innate immune responses. Previous studies have found that HBeAg inhibits the expression of TLR2 in cell lines and in vivo. However, the impact of bile acids on HBV infection remains obscure. Therefore, we set out to explore the effect of CDCA on innate immune molecules in HepG2. Cells were treated with DMSO or CDCA, which were simultaneously transfected with wild-type HBV plasmid pAAV/HBV1.2. The expression of TLR2 was significantly decreased in the group treated with CDCA (Figure 3A). On the contrary, the TLR3, TLR4, and TLR9 expression levels had no distinction. Furthermore, using the HBeAg nonsense mutation plasmid of pAAV/HBV1.2, there was no statistical difference in the expression of TLRs (Figure 3B). We argue that CDCA promotes the expression of HBeAg through nuclear receptor FXR and inhibits the expression of TLR2.
 renders aberrant toll-like receptor 2 (TLR2) expression by farnesoid X receptor (FXR) in HepG2. HepG2 cells were transfected with pAAV/HBV1.2 or HBeAg-null-pAAV/HBV1.2 and activated through FXR and chenodeoxycholic acid (CDCA). Total cellular RNA was isolated and analyzed by reverse-transcription (RT)-PCR. A, CDCA and FXR inhibited the expression of TLR2 in wild-type hepatitis B virus (HBV)-transfected HepG2 cells; B, CDCA and FXR did not inhibit the expression of TLR2 in HBeAg-null HBV transfected HepG2 cells. Three independent experiments were performed. * P < 0.05.")
Upregulation of hepatitis B e antigen (HBeAg) renders aberrant toll-like receptor 2 (TLR2) expression by farnesoid X receptor (FXR) in HepG2. HepG2 cells were transfected with pAAV/HBV1.2 or HBeAg-null-pAAV/HBV1.2 and activated through FXR and chenodeoxycholic acid (CDCA). Total cellular RNA was isolated and analyzed by reverse-transcription (RT)-PCR. A, CDCA and FXR inhibited the expression of TLR2 in wild-type hepatitis B virus (HBV)-transfected HepG2 cells; B, CDCA and FXR did not inhibit the expression of TLR2 in HBeAg-null HBV transfected HepG2 cells. Three independent experiments were performed. * P < 0.05.
4.6. Downregulation of HBeAg Results in Elevation of TLR2 in FXR-/- Mouse
Due to HBV transgenic mice serving as an immune tolerance model, we established a mouse model by hydrodynamic injection of pAAV/HBV1.2 in the immunocompetent mouse. As expected in HepG2 cells, the expression of HBeAg in serum was lower in FXR-/- mice than those in WT mice (Figure 4A). We further investigated whether FXR could regulate the HBV innate immune response in FXR-/- mice. Total RNA was extracted from the livers of mice, and RT-PCR analyzed innate immune molecules. The livers of FXR-/- mice showed a significant upregulation of TLR2 compared to WT mice. The levels of TLR3, TLR4, and TLR9 did not vary between the two groups of mice (Figure 4B and C).
 and toll-like receptor 2 (TLR2) in wild-type and farnesoid X receptor (FXR) knock-out mice. Serum specimens and livers were collected at days 0 and 7 after hydrodynamic injection of pAAV/HBV1.2. A, The expression of HBeAg in serum was detected by enzyme-linked immunosorbent assay (ELISA); B, The level of TLRs in the liver was analyzed using reverse-transcription (RT)-PCR at day 0; C, The level of TLRs in the liver at day 7. Five independent experiments were performed. At least four mice per group were analyzed. * P < 0.05.")
The negative correlation between hepatitis B e antigen (HBeAg) and toll-like receptor 2 (TLR2) in wild-type and farnesoid X receptor (FXR) knock-out mice. Serum specimens and livers were collected at days 0 and 7 after hydrodynamic injection of pAAV/HBV1.2. A, The expression of HBeAg in serum was detected by enzyme-linked immunosorbent assay (ELISA); B, The level of TLRs in the liver was analyzed using reverse-transcription (RT)-PCR at day 0; C, The level of TLRs in the liver at day 7. Five independent experiments were performed. At least four mice per group were analyzed. * P < 0.05.
5. Discussion
Chronic HBV infection is a significant public health problem. Furthermore, it is the leading cause of hepatitis, cirrhosis, and hepatocellular carcinoma worldwide (19). The mechanisms of chronic persistent HBV infection are unclear at present. Like a “stealth virus,” HBV invades the host’s immune surveillance (20). Hepatitis B e antigen is conserved in the hepatovirus family and secreted from infected hepatocytes as a co-protein of HBV but is not a structural protein of the virus (16). Infants born to serologically positive mothers often develop chronic hepatitis, showing that HBeAg exhibits some immunomodulatory functions (21). Activation of TLRs signaling contributes to the production of pro-inflammatory cytokines, such as IL6 and TNFα, which are conducive to viral inhibition (8). A previous study has proved that HBeAg inhibits the expression of TLR2 on hepatocytes in chronic HBV infection (6). Our study demonstrated that bile acid nuclear receptor FXR promotes the expression of HBeAg and inhibits the expression of TLR2 in vivo and in vitro.
The bile acids may inhibit immune response or induce immune tolerance in HBV chronic infection. The concentration of bile acids in serum highly rises during the process of acute HBV infection (22). Recent studies carried out in HBV-infected humanized mice have shown that HBV binds to its receptor NTCP, which is responsible for the uptake of bile acids into hepatocytes and promotes the synthesis of compensatory bile acids (15). If the concentration of bile acids is persistently elevated, it suggests that the progress of the disease would be chronic (22). Immunosuppression of bile acids may be attributed to the inhibition of monocyte activity assessed in terms of the production of monokines (23). In our opinion, impaired secretion of interleukins is directly involved in the immunosuppression of bile acids, which induce the production of HBV tolerogen.
Bile acids receptor FXR is part of the nuclear receptor superfamily of ligand-activated transcription factors, which play substantial roles in virus replication (24). As a transcription factor, it binds to FXR response elements as a monomer or a heterodimer with a retinoid X receptor (RXR) to regulate the expression of various target genes, including bile acid metabolism, inflammation, and viral and innate immune responses (25). The FXR response element in enhancer regions and the core promoter of HBV identified by Ramiere et al. is accountable for synthesizing viral RNA and DNA replication in transfected cell lines (26). Conversely, SHP is an FXR target activated by bile acids and inhibits the biosynthesis of HBV (27). Our study demonstrates that FXR increases the expression of HBeAg in vivo and in vitro.
Regulation of TLR2 expression in chronic HBV infection by HBeAg is partly due to the activity of bile acid receptor FXR. TLR2, also known as CD282, is a receptor for molecular patterns associated with bacterial, fungal, viral, and parasitic pathogenic pathogens (PAMPs). Toll-like receptor 2 (TLR2) signaling pathways inhibit HBV replication (28). On the contrary, HBeAg downregulates TLR2 and IL-1β-mediated responses (9, 29). Our study shows that in cell lines transfected with wild-type HBV and treated with bile acids, the expression of HBeAg is increased, and the expression of TLR2 is correspondingly declined. When using HBeAg-null HBV plasmid, the expression of TLR2 is no longer declined. Using a hydrodynamic injection HBV mouse model, HBeAg expression was decreased, and TLR2 was significantly increased in the FXR-/- mouse model, differently from HBV transgenic mice used as an immune tolerance model. It has been reported that HBeAg as a tolerogen may induce immune tolerance in infants and promote viral persistence during chronic HBV infection in adults (21). The mechanism of HBV chronic persistent infection may be partially explained through the FXR-HBeAg-TLR2 axis.
In conclusion, we investigated for the first time the role of the bile acid nuclear receptor FXR in HBV infection. Our data indicate that elevated serum bile acids may cause immune tolerance and lead to virus persistence in HBV-infected patients. This study suggests a potential clinical value for FXR antagonists in treating chronic HBV infection.
