1. Background
2. Objectives
3. Materials and Methods
3.1. Cell Culture
3.2. Transfection of cells and establishment of stable clones
3.3. Colony Formation Assay
3.4. Luciferase Assay
3.5. Extraction of RNA and Reverse Transcription-Polymerase Chain Reaction
| Gene | Name | Sequence (5’→3’) |
|---|---|---|
| DLEC1 | DLEC1LCF2 | CACGGAAGAGGCATCG |
| DLEC1LCR2 | ATAAGCCGGAAGTAGTGT | |
| Cyclin A2 | CA2F | TTTGGGGCCTCCTGTCTG |
| CA2R | TGCTTCAGACTCCACGGG | |
| Cyclin B1 | CB1F | TGCTGCAACCTCCAAGCC |
| CB1R | GGCACCAGCATAGGTACC | |
| Cyclin D1 | CD1F | CCATGGAACACCAGCTCC |
| CD1R | AAGACCTCCTCCTCGCAC | |
| Cyclin D3 | CD3F | GCTGCTGTGTTGCGAAGG |
| CD3R | CATGGCCAGGGGGAAGAC | |
| Cyclin E1 | CE1F | ACAGGGATGCGAAGGAGC |
| CE1R | AGGGGTCTGCACAGACTG | |
| p21 | P21F | CACCGAGGCACTCAGAGGA |
| P21R | TCGAAGTTCCATCGCTCACG | |
| SP1 | SP1F | TTGTCCCTCAGCTGCCAC |
| SP1R | GGCTGTGAGGTCAAGCTC | |
| SP3 | SP3F | ATGGCTGCCTTGGACGTG |
| SP3R | AATCACCTGTCGCTCCGG | |
| AP2α1 | AP2α1F | ATATCCGTTCACGCCGATCC |
| AP2αR | GGGGGGAAGTATGGGGGCTG | |
| AP2α2 | AP2α2F | TCGTTACCCCAGACTCTTCG |
| AP2 α R | GGGGGGAAGTATGGGGGCTG | |
| AP2γ | AP2γF | ACTGGCGACTGTTTTGGGGG |
| AP2γR | GGGGGAAAGTAGGGTGGCGG | |
| AP2ε | AP2εF | AGCTACCGCACCGTGACCTC |
| AP2εR | GGCGGGAAGTAGGGCGGCTG |
aReal-time RT-PCR: Conventional RT-PCR.
3.6. Extraction of Protein and Western Blotting
3.7. Flow Cytometry (FACS) and Cell Proliferation Assay
3.8. Statistical Analysis
4. Results
4.1. The Transcriptional Regulatory Potency of DLEC1

A, Schematic illustration of the full-length of DLEC1 ORF and its four fragments used in this study. Numbers with arrows denote the boundary nucleotide position in DLEC1 DNA fragments (top); schematic representation of the four peptides encoded by DLEC1 DNA fragments. Numbers with bars depict boundary amino acid residues in DLEC1 protein fragments (bottom); B, Relative luciferase activities affected by DLEC1 and its fragments. HCT116 cells were transiently co-transfected with firefly luciferase reporter plasmid, GAL4-DLEC1 (or fragments) construct and Renilla luciferase plasmid. Relative luciferase activity of each sample was expressed as firefly luciferase activity divided by the corresponding Renilla luciferase activity to correct for transfection efficiency and subsequently standardized to that from the empty vector control (GAL4). All data are presented as mean ± Standard Error (SE) and are representative of one of at least two independent experiments performed in triplicates.
4.2. Characterization of DLEC1-Overexpresing Stable Clones in the HCT116 Cell Line

Stable clones of DLEC1 over-expression were generated by transfection of pcDNA31DLEC1 construct in HCT116. On G418 selection, positive stable clones were verified for DLEC1 expression level by A, Real-time RT-PCR and B, Western Blotting. The relative expression of DLEC1 was expressed as DLEC1 mRNA level divided by GAPDH mRNA level and subsequently normalized against that from the pcDNA31 vector control. DLEC1 over-expression was able to suppress cell proliferation as shown by C, representative colony formation; D, quantification of colony formation and E, MTT assay. The initial plated cell numbers are indicated below. All data are presented as mean ± SE and are one representative of at least two independent experiments performed in triplicates. *, P < 0.05.
| Variables | Controls | DLEC1-Transfected Stables | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 4 | 3 | 4 | 5 | 6 | 7 | 10 | 11 | 15 | |
| Sub-G1 | 16.3 | 11.6 | 7.1 | 10.1 | 8.2 | 13.1 | 6.6 | 10.1 | 12.1 | 36.2 |
| G1 | 46.3 | 41.0 | 57.6 | 56.7 | 54.0 | 66.4 | 57.8 | 56.7 | 55.5 | 48.3 |
| S | 19.8 | 26.1 | 22.0 | 18.1 | 22.3 | 12.2 | 18.2 | 18.1 | 17.2 | 8.1 |
| G2/M | 17.6 | 21.3 | 12.3 | 15.1 | 15.5 | 8.3 | 17.4 | 15.1 | 15.2 | 7.4 |
4.3. The Role of p21 in G1 Arrest

Cultured stable cells were harvested and RNA or protein was prepared for A, conventional RT-PCR; B, Real-time RT-PCR or C, Western blotting. GAPDH or β-actin was used as a loading control for PCR or Western blot analysis, respectively. The relative expression of p21 was expressed as the p21 mRNA level divided by GAPDH mRNA level and subsequently normalized against that from the pcDNA31 vector control.
4.4. The Role of AP-2α2 in Mediation of DLEC1-Regulated Cancer Cell Growth

Stable cells were cultured for extraction of RNA used in conventional RT-PCR analyses with indicated genes. GAPDH was used as an internal control for PCR.

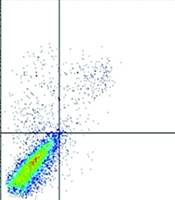
A, Down-regulation of AP-2α2 by siRNAs against AP-2α2; B, relative cellular proliferation; C, cell cycle analyses; and D, alteration of cells in sub-G1 and G1 fractions after the down-regulation of AP-2α2. Stable cells of three clones were transfected with AP-2α2 siRNAs and cells were harvested for analysis after 72 hours (or as indicated). For fluorescence activated cell-sorting (FACS) analysis, transfected cells were harvested, fixed, treated with RNase, and stained by PI. Cells in the sub-G1 phase were regarded as apoptotic cells. The numbers in insets of C indicate sub-G1 (upper) or G1 (lower) percentage of cells. The numbers in D are the comparison results of sub-G1 or G1 in AP-2α2 knockdown samples with respective SCR samples in each stable clones. All data are presented as mean ± SE and are one representative of at least two independent experiments performed in triplicates. *, P < 0.05; **, P < 0.01.