





1. Background
Non-alcoholic fatty liver disease (NAFLD), often linked to metabolic syndrome and diabetes, can escalate to nonalcoholic steatohepatitis (NASH), leading to cirrhosis and potentially hepatocellular carcinoma, influenced by dietary habits, environmental, and genetic factors (1, 2). While the precise mechanisms underlying its pathogenesis remain elusive, it is evident that inflammatory mediators play a substantial role (3). The rising prevalence of NAFLD is accompanied by an increase in patient numbers and healthcare costs, imposing a significant economic burden on society (4). In terms of clinical applications, Bifidobacterium bifidum has been shown to have a positive effect on the treatment of chronic diarrhea, antibiotic-associated diarrhea, and constipation. In addition, bifidobacteria may also play a role in tumor prevention and treatment, liver protection, and prevention of hypertension and atherosclerosis.
Chronic obstructive pulmonary disease (COPD) is a chronic inflammatory lung disease characterized by irreversible progressive airflow limitation. It represents a significant challenge to global health, with the global Initiative for chronic obstructive lung disease (COLD) emphasizing that COPD is one of the three leading causes of death worldwide (5, 6). The COPD risk factors include exposure to harmful particles or gases, predominantly affecting low-and-middle-income countries with aging populations (7). The disease's pathogenesis, while not fully elucidated, involves inflammatory factors that contribute to lung damage, exacerbating the global economic burden (8).
Patients with COPD have a higher prevalence of severe hepatic steatosis than the general population (9). Patients with NAFLD frequently exhibit comorbid hyperglycemia, dyslipidemia, and elevated Body Mass Index (BMI), which are established risk factors for COPD (9). Although NAFLD and COPD are linked, it appears that the subject of previous investigation, relevant genetic studies are still limited, and the relevant gene regulatory network has not been fully established, which requires further exploration.
2. Objectives
This project is based on a bioinformatics approach to identify the co-expressed genes of NAFLD and COPD, employing systems biology analytical methods to further elucidate their connections and preliminary experimental validation of the screened key genes by setting up NAFLD and COPD models in vitro. The central genes identified in this investigation may serve as a focal point for future research, and the molecular mechanisms and signaling pathways delineated here could provide valuable insights into the interplay between COPD and NAFLD.
3. Methods
3.1. Materials
HepG2 and BEAS-2B cell lines were sourced from the Institute of Basic Medical Sciences, Peking Union Medical College, China. Cell culture was performed in glucose-rich DMEM supplemented in accordance with the manufacturer's instructions, fetal bovine serum (Gibco, USA), and rabbit monoclonal antibodies against COX-2, cycle protein-dependent kinase inhibitor 1A (CDKN1A), formyl peptide receptor 1 (FPR1), ficolin-1 (FCN1), CLEC4D, and β-actin (Abcam, UK) were used.
3.2. Microarray Data Collection
Microarray datasets related to NAFLD, COPD, and healthy subjects were retrieved from the GEO database. Specifically, a genetic overview of 11 individuals with NAFLD and 7 control individuals with NAFLD controls are included within the GSE63067 dataset. In the GSE37768 dataset, the relevant gene expression data of 20 COPD patients and 18 controls were analyzed, of which 9 non-smoking healthy people were selected as controls.
3.3. Identifying Genes Variably Represented in Non-alcoholic Fatty Liver Disease and Chronic Obstructive Pulmonary Disease
Differentially expressed genes (DEGs) were identified using the limma package in the R software between patients with NAFLD and COPD and their respective controls (10, 11). The criteria for selection in NAFLD as well as COPD datasets were set at P < 0.05 and |log2 FC| > 0.5. This dual-criteria approach balances sensitivity and specificity to capture biologically relevant changes while minimizing false negatives. The choice of |log2FC| > 0.5 is supported by prior studies in NAFLD and COPD, where this threshold has been shown to effectively identify genes with moderate but biologically significant expression changes (12-14). Additionally, the stringent P-value cutoff of < 0.05 further ensures the robustness of our findings. After shortlisting, two sets of DEGs were established and analyzed using the Venn diagram tool available online to discover overlaps between the two diseases. Intersecting genes were obtained for follow-up analysis.
3.4. Common Differentially Expressed Gene Functional Enrichment Analysis
We analyzed the molecular features, biological processes, and cellular constituents of intersecting genes using GO and KEGG pathway enrichment analyses in the R software (15). We defined P < 0.05 as statistically significant.
3.5. Identification of Key Genes and Construction of PPI Networks
To examine the interactions of the common genes identified, a PPI network was set up using the Interacting Gene Search Tool (STRING) release 11.0 (16). The lowest interaction threshold was set at medium confidence (0.400) to establish statistical significance, and the maximum number of tier 1 and 2 interactions was limited to 10. In the resulting network, nodes represent proteins, while edges indicate interactions between them.
3.6. Establishment of Non-alcoholic Fatty Liver Disease and Chronic Obstructive Pulmonary disease Cell Models
The NAFLD model was constructed by treating HepG2 cells with 1
3.7. Oil Red O Staining for Cellular Lipid Accumulation Detection
Cells treated as described in "2.6" were washed three times with PBS. Cells were fixed for 30 minutes with 1 mL paraformaldehyde per well. They were then stained with 400 μL Oil Red O for 20 minutes in the dark, followed by washing. Finally, cells were rinsed with PBS and imaged under an optical microscope. A UV-Vis spectrophotometer was used to measure the absorbance of the extracted solution at a wavelength of 510 nm.
3.8. Detection of COX-2, Cycle Protein-Dependent Kinase Inhibitor 1A, Formyl Peptide Receptor 1, Ficolin-1, CLEC4D Protein Expression by Western Blot
After the cells were treated for 24 hours, the protein lysate was used to lyse the cells to extract gross proteins from the cells of each group. The concentration of protein was then determined using the Bicinchoninic Acid Assay. Following quantification, the proteins were subjected to vertical SDS-PAGE, transferred to a moist PVDF membrane, and blocked for 20 minutes. The membranes were then incubated with primary antibodies (1:5000) at 4°C overnight, followed by three rounds of TBST washing. Subsequently, they were incubated with a secondary antibody in the dark for 1 hour and detected using an infrared fluorescent scanning and imaging system. The results were analyzed using Image J software.
3.9. Statistical Analysis
Statistical analyses were performed using SPSS version 22.0. For comparisons between two independent groups, independent-samples t-tests were used. When data did not meet the assumptions of normality, the nonparametric Mann-Whitney U test was applied. For multiple group comparisons, one-way ANOVA was conducted, followed by Tukey's post-hoc test for pairwise comparisons to control for Type I error. Correlation analyses were performed using Pearson's correlation for normally distributed data and Spearman's rank correlation for non-normally distributed data. Receiver operating characteristic (ROC) curves were generated in GraphPad Prism to compare area under the curve (AUC) values. Multivariate regression models were used to address potential confounders. Results are presented as mean ± standard deviation (X̄ ± SD). A P-value of less than 0.05 was considered statistically significant, * indicates a significant difference..
4. Results
4.1. Differential Expression Analysis of Differentially Expressed Genes in Non-alcoholic Fatty Liver Disease and Chronic Obstructive Pulmonary Disease
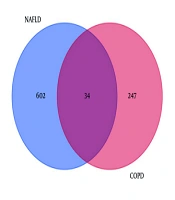
Heatmaps of the gene expression profiles of NAFLD and COPD are shown in Figure 1, with 636 and 281 DEGs obtained under the condition of P < 0.05. The 34 genes that overlap and are common to the two diseases were identified using the online Venn diagram tool (Figure 2). Detailed information on the common genes is presented in Appendix 1 in Supplementary File, while Figure 3 displays specific information regarding the analysis process.
 compared to the control group; B, differential gene of chronic obstructive pulmonary disease (COPD) compared to the control")
Differential genes heat map of diseases – A, differential gene of non-alcoholic fatty liver disease (NAFLD) compared to the control group; B, differential gene of chronic obstructive pulmonary disease (COPD) compared to the control
 and chronic obstructive pulmonary disease (COPD) differential genes")
Venn diagram of non-alcoholic fatty liver disease (NAFLD) and chronic obstructive pulmonary disease (COPD) differential genes

Analysis flow chart
4.2. GO and KEGG Enrichment Pathway Analysis
Enrichment analysis for functions and KEGG pathways of the 34 common genes in NAFLD and COPD was carried out using a P-value threshold of < 0.05. The main changes in the GO Biological Processes (BP) are as follows: Activation of neutrophils involved in immune responses, neutrophil-driven immunity, and the regulation of innate immune response. Changes in Cellular Components (CC) were mainly focused on fibronectin-rich granule body membranes, the inner lumen, and the lumen of the tertiary granule. In addition, in the molecular function (MF) category, significant changes were observed in glycosylation end product receptor binding, pattern recognition receptor activity, and carbohydrate binding modules. Specifically, KEGG pathway changes focused on viral proteins interacting with cytokines and their corresponding receptors, the CLR-mediated signaling pathways, and cytokine-mediated cell-cell communication interactions (Figure 4).
; D, KEGG signaling pathways")
Common gene function enrichment information – A, biological process; B, cell component; C, molecular function (MF); D, KEGG signaling pathways
4.3. PPI Network Analysis and Core Gene Selection
To clarify the interrelationships of shared genes, we set up a PPI network. Proteins with interactions with other proteins were identified as more critical genes. These serve as the core nodes in the protein interaction network, with the precise degree of connectivity indicating the central gene's role, as shown in Figure 5. Less information was found about the interacting proteins, so information mining was used to expand the network. Finally, the group selected proteins with interactions greater than 2 as key targets for further research.

Inter-gene interactions – A, differential gene protein interaction network; B, differential protein with interaction; C, expanded interacting protein
To verify the predictive validity of the four central genes (CDKN1A, FPR1, FCN1, CLEC4D) based on the analyses, ROC curves were created using the results from the above analyses, and the AUC for transcriptional activity levels was determined in the NAFLD and COPD datasets. The outcomes of the ROC analysis for NAFLD and COPD are shown in Figure 6. The AUCs for CDKN1A, FPR1, FCN1, and CLEC4D among NAFLD patients and healthy controls were 0.767 (95% CI, 0.573 - 0.96; P < 0.05), 0.7 (95% CI, 0.5 - 0.9; P = 0.079), 0.656 (95% CI, 0.445 - 0.866; P = 0.72), and 0.9 (95% CI, 0.787 - 1; P < 0.001), respectively. The AUCs for the four central genes in COPD were 0.772 (95% CI, 0.584 - 0.959; P < 0.05), 0.741 (95% CI, 0.549 - 0.933; P < 0.05), 0.802 (95% CI, 0.623 - 0.982; P < 0.05), and 0.796 (95% CI, 0.62 - 0.973; P < 0.05), respectively.
 curve of hub genes – A, ROC information of hub genes in chronic obstructive pulmonary disease (COPD); B, ROC information of non-alcoholic fatty liver disease (NAFLD)")
Receiver operating characteristic (ROC) curve of hub genes – A, ROC information of hub genes in chronic obstructive pulmonary disease (COPD); B, ROC information of non-alcoholic fatty liver disease (NAFLD)
4.4. Establishment of Non-alcoholic Fatty Liver Disease and Chronic Obstructive Pulmonary Disease Cell Models
Oil Red O, a lipophilic dye that can dissolve in fats, specifically stains neutral fats like triglycerides in cells red. Hematoxylin, a basic natural dye, can stain cell nuclei (18). As shown in Figure 7A and B, the lipid content in the NAFLD model group was markedly higher. The TG level in the NAFLD model group was significantly higher than in the control group (in Figure 7C). The COPD is accompanied by severe inflammatory responses. COX-2 can further transform arachidonic acid into eicosanoids, including prostaglandins. Research reveals that COX-2 expression is associated with the occurrence or amplification of inflammation (19). Therefore, COX-2 is chosen here to verify the successful establishment of the COPD model. Compared with the control group, COX-2 expression in A549 cells in the COPD model group was significantly increased (P < 0.001), as shown in Figure 7D and E, indicating a marked inflammatory response in the cells.
 and chronic obstructive pulmonary disease (COPD) cell models – A, oil Red O staining of cells in the NAFLD group; B, oil Red O staining absorbance comparison; C, comparison of triglyceride levels in normal and NAFLD cells; D, COX protein gel; E, western blot statistical chart – compared with control group, * P < 0.05, ** P < 0.01, *** P < 0.001, n=3 (* indicates a significant difference).")
Establishment of non-alcoholic fatty liver disease (NAFLD) and chronic obstructive pulmonary disease (COPD) cell models – A, oil Red O staining of cells in the NAFLD group; B, oil Red O staining absorbance comparison; C, comparison of triglyceride levels in normal and NAFLD cells; D, COX protein gel; E, western blot statistical chart – compared with control group, * P < 0.05, ** P < 0.01, *** P < 0.001, n=3 (* indicates a significant difference).
4.5. Detection of Cycle Protein-Dependent Kinase Inhibitor 1A, Formyl Peptide Receptor 1, Ficolin-1, and CLEC4D Protein Expression by Western Blot
The CDKN1A, FPR1, FCN1, and CLEC4D protein expression was detected by Western blot. The NAFLD model group indicated a pronounced enhancement in CDKN1A, FPR1, FCN1, and CLEC4D protein expression compared with the blank group (Figure 8A). Similarly, the COPD model group exhibited a considerable enhancement in CDKN1A, FPR1, FCN1, and CLEC4D protein expression compared to the blank group, with CLEC4D protein expression also significantly increased (Figure 8B - E). All the differences were statistically significant (P < 0.05). This suggests that CDKN1A, FPR1, FCN1, and CLEC4D are likely to play a crucial role in the initiation and progression of NAFLD and COPD.
, formyl peptide receptor 1 (FPR1), <i>ficolin-1</i> (<i>FCN1</i>), <i>CLEC4D</i> – A, each protein strip graph; B, <i>CDKN1A</i> protein expression; C, FPR1 protein expression; D, <i>FCN1</i> protein expression; E, <i>CLEC4D</i> protein expression – compared with control group, * P < 0.05, n = 3")
The protein expression of cycle protein-dependent kinase inhibitor 1A (CDKN1A), formyl peptide receptor 1 (FPR1), ficolin-1 (FCN1), CLEC4D – A, each protein strip graph; B, CDKN1A protein expression; C, FPR1 protein expression; D, FCN1 protein expression; E, CLEC4D protein expression – compared with control group, * P < 0.05, n = 3
5. Discussion
Both NAFLD and COPD are quite common around the world. Previous studies have shown that patients with COPD are more likely to have problems with NAFLD (20). The progressive increase in risk with longer disease duration necessitates further investigation. In this study, we explored NAFLD and COPD datasets from GEO and identified 34 shared DEGs. We conducted GO and KEGG pathway enrichment analyses and built a PPI network to pinpoint key genes within the common DEGs, shedding light on the molecular mechanisms and potential early targets for preventing disease progression in both conditions. We selected four key genes to evaluate their diagnostic potential in patients with NAFLD and COPD (P < 0.05). It is possible that these genes may serve as valuable predictors for the risk of developing COPD and NAFLD.
Cell CDKN1A is a biomarker of cell cycle arrest and premature senescence, and it has been found that CDKN1A expression is higher in the peripheral lungs of patients with COPD, which may be associated with muscle dysfunction (21). Skeletal muscle atrophy is a frequent complication in COPD patients (22). The CDKN1A was upregulated in the muscle tissue of COPD patients compared to healthy controls (23). The COPD patients show altered levels of CDKN1A, indicating its potential role in the development of COPD (19). The level of CDKN1A protein was also found to change significantly during the progression of NASH to the development of HCC (24), and a reasonable hypothesis has been put forward for the effect of CDKN1A in NAFLD, suggesting that the CDKN1A rs762623 variant may help protect hepatocytes from advanced fibrosis by mitigating their senescence (25). In COPD, CDKN1A is elevated in peripheral lungs and muscle tissue, potentially contributing to muscle dysfunction and skeletal muscle atrophy (26). In NAFLD, CDKN1A levels change during NASH progression to HCC, with a variant possibly protecting hepatocytes from fibrosis (27, 28).
The FPR1 is a member of the GPCR superfamily. It is predominantly expressed in mammalian leukocytes and plays a crucial role in inflammatory responses, as well as in the regulation of brain homeostasis (29). When hepatic necrosis was induced in mice, a rapid accumulation of neutrophils was found, but when FPR1 was inhibited, the migration of neutrophils to the area of hepatic necrosis was significantly reduced (30). The FPR1/FPR2 heterodimerization triggers a delayed and induced pro-inflammatory response to the JNK pathway with neutrophil apoptosis (31). The FPR1 has been shown to be an important receptor in COPD, as the gene alteration contributes to protection against cigarette smoke-induced emphysema formation in a mouse model, consistent with other findings that FPR1 expression is increased in neutrophils from COPD patients with high levels of dyspnea (32). The FPR1, a GPCR family member crucial for inflammatory responses, shows altered expression in COPD patients and mouse models, indicating its role in cigarette smoke-induced emphysema and neutrophil regulation (32). In NAFLD, FPR1 inhibition reduces neutrophil migration to hepatic necrosis sites, suggesting its involvement in liver inflammation (33).
The FCN1, encoded by the FCN1 gene, is synthesized in the bone marrow and is present in type II alveolar epithelial cells, granulocytes, and monocytes (34). This protein contains collagen-like domains that interact with mannose-binding lectin-associated serine protease (MASP), thereby triggering the complement lectin pathway (LP) cascade upon target binding (35). Notably, the interaction of M-fibrillar collagen with natural killer (NK) cells differs from that with activated T cell subsets. While NK cells interact directly with M-fibrillar collagen, T cell subsets engage via specific ligand binding sites in the FBG structural domain, bridging adaptive and innate immunity (36). Elevated serum levels of M-ficolin have been linked to exacerbated inflammation, potentially leading to poorer outcomes in pediatric pneumonia (37).
Recent multi-omics research has identified FCN1 as a key gene in NAFLD, implicating it in disease development, particularly in regulating fat accumulation and liver inflammation (38). FCN1 also appears to influence immune cell infiltration, possibly through ceRNA regulation, which is critical in atherosclerosis progression (39). In NAFLD, FCN1 activates the complement system via the LP by recognizing and binding to specific carbohydrates and injured cells, such as apoptotic cells. This binding activates associated serine proteases like MASP, which then cleave C4 and C2 to form the C3 convertase. The subsequent cleavage of C3 into C3a and C3b initiates a cascade of complement activation reactions, contributing to inflammation and liver injury. For example, a study found that FCN1 is involved in humoral immune responses, complement activation, and phagocytosis in PIBD (40). Our study further solidifies FCN1's involvement in NAFLD-related fat accumulation, liver inflammation, and immune cell infiltration.
In COPD's pathologic process, macrophages mainly polarize to the M1 type. M1 macrophages secrete pro-inflammatory cytokines like IL-6, TNF-α, and IL-1β, recruiting other immune cells and worsening pulmonary inflammation (41). CLEC4D (Member 4 of the C-type lectin family), predominantly expressed in macrophages and myeloid cells, plays a role in the body's defense against mycobacterial infections (37). In humans, CLEC4D gene polymorphisms with reduced expression have been associated with increased susceptibility to tuberculosis (42). CLEC4D also impacts chronic diseases, being implicated in causing type 1 diabetes mellitus (T1D) and contributing to adipose tissue fibrosis through the interaction between adipocytes and macrophages (41). Previous studies link CLEC4D polymorphisms to tuberculosis susceptibility and T1D development (42). Our research adds to this by showing its involvement in adipose tissue fibrosis and macrophage responses to viral infections.
CLEC4D likely regulates macrophage polarization in COPD through multiple mechanisms (43). It may recognize components in cigarette smoke or lung DAMPs, activating macrophage signaling pathways like NF-κB to promote M1 polarization (44). Additionally, CLEC4D may interact with other macrophage receptors or signaling molecules, modulating intracellular signaling and influencing polarization-related gene expression to drive the shift to the M1 phenotype (41). However, the exact molecular mechanisms and signaling transduction require further study.
In COPD's pathologic process, macrophages mainly polarize to the M1 type. M1 macrophages secrete pro-inflammatory cytokines like IL-6, TNF-α, and IL-1β, recruiting other immune cells and worsening pulmonary inflammation. CLEC4D likely regulates this polarization through multiple mechanisms. It may recognize components in cigarette smoke or lung DAMPs, activating macrophage signaling pathways like NF-κB to promote M1 polarization. Additionally, CLEC4D may interact with other macrophage receptors or signaling molecules, modulating intracellular signaling and influencing polarization-related gene expression to drive the shift to the M1 phenotype. However, the exact molecular mechanisms and signaling transduction require further study (45).
In this study, COPD and NAFLD datasets were extracted from GEO, and a comparative analysis between patient and normal samples identified 34 shared genes. The GO and KEGG enrichment analyses were then conducted to identify immune-related biological functions and pathways. Key genes are emerging as central to these mechanisms. The pivotal genes CDKN1A, FPR1, FCN1, and CLEC4D were selected by constructing a PPI network. We identified key genes, and their diagnostic potential was validated through data analysis with SPSS. The important roles of the four key genes (CDKN1A, FPR1, FCN1, and CLEC4D) in the disease were preliminarily verified by in vitro establishment of NAFLD and COPD models. Our research outcomes might offer promising therapeutic targets for the management and prevention of NAFLD and COPD.
However, our study has certain limitations. First, the DEGs identified via bioinformatics could potentially serve as predictive biomarkers for NAFLD and COPD development. Yet, the lack of in vitro validation and clinical cohort studies restricts the confirmation and generalizability of these findings. Second, the analyzed samples come from diverse ethnic groups, so the applicability of these results may be limited to specific populations and may not necessarily apply to all demographic groups. Moreover, the sample sizes are relatively small, which may lead to biases and limit the representativeness of the broader population. Additionally, our study did not specifically examine potential biases related to age or gender differences, which could further influence the generalizability of our findings. Therefore, further studies are needed in other populations, with larger sample sizes, and more detailed consideration of ethnic, age, and gender factors to validate our results and ensure their broader applicability.
In our study, several limitations should be acknowledged. We did not account for batch effects in GEO datasets. Such batch effects, arising from variations in experimental conditions, processing times, or technical factors, might have influenced our analysis results. Additionally, the risk of cell line misidentification was not considered. Misidentified or cross-contaminated cell lines could lead to inaccurate findings and affect the reliability of our conclusions. Moreover, while HepG2 and A549 cells provided molecular insights into NAFLD and COPD, they do not fully mirror human pathophysiology. Future studies should use patient-derived samples like liver/lung biopsies to boost clinical relevance.
Also, the sample sizes in GSE63067 and GSE37768 were small. Although the high power and significant effect sizes suggest robust results, the limited samples may restrict generalizability. Despite these limitations, this study reveals key shared molecular mechanisms between NAFLD and COPD and pinpoints genes for further study. These insights enhance our understanding of disease pathogenesis and lay a foundation for future research. In future studies, we will implement stricter quality control measures and validation steps to address the aforementioned issues and minimize their potential impact.
5.1. Conclusions
The results of this study show that CDKN1A, FPR1, FCN1, and CLEC4D are key genes associated with NAFLD and COPD. The key genes screened have been preliminarily verified in cell experiments and are likely to contribute significantly to the pathogenesis and progression of NAFLD and COPD, thus providing a new theoretical basis for the occurrence of these diseases.
