











1. Background
Although hepatitis B virus (HBV) infection is preventable through vaccination, it continues to be a major cause of liver-related morbidity and mortality worldwide (1). According to a 2022 report by the World Health Organization (WHO), approximately 254 million people live with chronic HBV infection, which leads to around 1.1 million deaths annually (2). In Turkey, the seroprevalence of hepatitis B is reported to range from 2% to 8% (3). A crucial marker of a positive prognosis in chronic HBV infection is the loss of hepatitis B surface antigen (HBsAg) (4). However, HBsAg clearance is infrequent, primarily due to the persistence of covalently closed circular deoxyribonucleic acid (cccDNA) within hepatocytes. Spontaneous HBsAg loss is estimated to occur at an annual rate of approximately 0.5 - 1% (5). Recent studies have focused on identifying important clinical and viral factors that may help predict the spontaneous clearance of HBsAg. These factors encompass host-related variables such as gender, age, alanine aminotransferase (ALT) levels, presence of fatty liver or cirrhosis, as well as viral features including hepatitis B e antigen (HBeAg) status, HBV deoxyribonucleic acid (DNA) viral load, serum HBsAg levels, HBV genotype, and co-infection with hepatitis C virus (HCV) (6-11). Although these factors offer valuable information, accurately predicting HBsAg loss continues to be challenging. The diverse immune responses seen in chronic HBV patients are thought to be partly due to genetic variability among individuals (12).
Tremendous efforts have identified various host genetic variants influencing HBV infection susceptibility and outcomes, including polymorphisms in human leukocyte antigen (HLA) genes, cytokines, toll-like receptors (TLRs), microRNAs, the sodium taurocholate cotransporting polypeptide (NTCP, the HBV receptor), and vitamin D-related genes. A significant portion of genetic research has concentrated on cytokines and chemokines involved in modulating the immune response against HBV (13). Among these molecules, CXCL10 — also known as interferon gamma (IFN-γ)-inducible protein-10 (IP-10) — has gained attention for its involvement in HBV disease mechanisms. Research indicates that IP-10 facilitates the recruitment of leukocytes from circulation to the liver and stimulates natural killer (NK) cells to produce IFN-γ, contributing to the non-cytolytic clearance of intrahepatic cccDNA (14, 15). Additionally, the G-201A polymorphism located in the promoter region of the IP-10 gene has been associated with disease progression in patients with chronic HBV, likely through its effect on regulating IP-10 expression (16). The polymorphism known as G-201A in Human Genome 19 is specified as -135G/A in Human Genome 38 [(NM_001565.4) c.-135C>T]. Previous studies have examined the relationship between the c.-135C>T polymorphism and disease progression in hepatitis B and C (17-20); however, its direct association with HBsAg loss has not yet been explored.
2. Objectives
To our knowledge, this is the first study to directly investigate the relationship between IP-10 promoter polymorphisms and spontaneous HBsAg clearance, aiming to elucidate a potential genetic mechanism underlying functional cure in chronic HBV infection. Based on the existing data, our study hypothesizes that polymorphisms within the IP-10 promoter region could influence the immune response and HBsAg clearance by modulating IP-10 expression. In addition to this polymorphism, we also aimed to investigate other variants in the IP-10 promoter region potentially associated with HBsAg clearance through sequencing analysis.
3. Methods
3.1. Study Population and Design
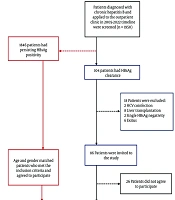
This study was conducted as a single-center, observational case-control investigation. Patients included were followed at the Department of Infectious Diseases and Clinical Microbiology, Faculty of Medicine, Cukurova University. A retrospective screening was performed on 1,950 chronic HBV patients who attended the Infectious Diseases and Clinical Microbiology Outpatient Clinic between July 2005 and December 2022. A total of 104 patients with chronic HBV infection who experienced HBsAg loss during follow-up were identified. The inclusion criteria for the study were: Age 18 years or older, diagnosis of chronic HBV infection defined by HBsAg positivity lasting at least 6 months, follow-up at the Infectious Diseases and Clinical Microbiology Outpatient Clinic for a minimum of 1 year, and documentation of at least two consecutive HBsAg negative results at 6-month intervals. Exclusion criteria included acute HBV infection, coinfection with human immunodeficiency virus (HIV) or HCV, liver transplantation, cirrhosis, hepatocellular carcinoma, and patients with transient HBsAg loss. The control group consisted of chronic HBV patients who remained HBsAg positive, met the inclusion criteria, had a similar age and gender distribution as the HBsAg loss group, and consented to participate in the study. These patients were selected from those who applied to the outpatient clinic since the start of the study. A total of 120 patients were included in the study, comprising 60 cases and 60 controls (Figure 1). Due to unsuitable Sanger sequencing results, polymorphism analysis could not be performed for a total of five patients — three from the case group and two from the control group. Peripheral blood samples were collected in vacuum tubes (BD Vacutainer®, England) containing 0.5 molar (M) ethylenediaminetetraacetic acid dipotassium (K2EDTA) (5.4 mg) and processed at the Chest Diseases Molecular Research Laboratory within the Biotechnology Center. The samples were stored at +4°C until DNA extraction. Genomic DNA was isolated, and primers targeting the IP-10 promoter region were designed for mutation analysis. While the primer design targeted the promoter region of the IP-10 gene, sequencing unexpectedly extended into exon 1, intron 1, and exon 2, allowing these regions to be analyzed as well. Based on a comprehensive search of the Genome-Wide Association Studies (GWAS) database and relevant literature, three variants were identified within these regions (c.-135C>T, c.85C>T, and c.83T>G). Sequence data were analyzed specifically with respect to these three variants.
.")
Flowchart of participant selection in this study (Abbreviations: HBV, hepatitis B virus; HBsAg, hepatitis B surface antigen; HCV, hepatitis C virus).
3.2. Isolation of Genomic Deoxyribonucleic Acid from Blood Samples
Blood samples collected from 120 patients in EDTA tubes were centrifuged after the addition of Fixol, and the white layer containing leukocytes was separated. Genomic DNA was isolated using the GeneAll® ExgeneTM Blood SV Mini Kit according to the manufacturer’s protocol. The purity and concentration of the extracted DNA were assessed using a NanoDrop spectrophotometer, and samples with absorbance ratios of 260/280 nm between 1.8 and 2.0, and 260/230 nm between 2.0 and 2.2 were included in the study.
3.3. Mutation Analyses
A 942-nucleotide segment covering the promoter, exon 1, exon 2, and intron 1 regions of the CXCL10 gene (NM_001565.4) was amplified using conventional polymerase chain reaction (PCR) with the forward primer (5′-3′) GAGGAGCAGAGGGAAATTCCG and reverse primer (5′-3′) CGTGGACAAAATTGGCTTGC. The PCR products were sequenced using Sanger sequencing on an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The sequencing data were analyzed with CLC Genomic Workbench 24 (Qiagen, Hilden, Germany) by aligning to the GRCh38/hg38 reference genome based on the CXCL10 gene (NM_001565.4). During analysis, primer binding sites, the first 20 and last 30 nucleotides, regions with a Phred quality score below 20, and sequences with background noise exceeding 20% were excluded.
3.4. Statistical Methods and Analyses
The required sample size was estimated using G*Power version 3.1.9.7. Based on this calculation, a minimum of 54 participants per group (total n = 108) was needed to achieve 80% statistical power with a significance level (α) of 0.05. The data were analyzed statistically using the Statistical Package for the Social Sciences (SPSS) version 25.0. Continuous variables were expressed as mean ± standard deviation or median with interquartile range (25th - 75th percentile) where appropriate, while categorical variables were presented as counts and percentages. The chi-square test was used to compare categorical variables. The Shapiro-Wilk test was applied to assess the normality of the data distribution. For normally distributed variables, the Independent Student’s t-test was used, whereas the Mann-Whitney U test was applied for non-normally distributed variables in comparisons between two groups. Statistical significance was defined as P < 0.05 for all analyses.
4. Results
4.1. Characteristics of Patients
The study included a total of 120 patients, with 60 individuals in the case group (HBsAg loss) and 60 in the control group (HBsAg positivity). Among 1,950 chronic HBV patients evaluated between 2005 and 2022, the overall rate of HBsAg loss was 5.33%. There were no statistically significant differences between the case and control groups regarding gender, age, age at diagnosis, Body Mass Index, alcohol consumption, or smoking status (P > 0.05). The mean age at the time of HBsAg loss was 53.2 ± 11.3 years, and 85% of patients experienced HBsAg loss after the age of 40. The average duration until HBsAg seronegativity was 15.0 ± 7.3 years, with 65% (n = 39) of patients becoming seronegative after more than 10 years. Table 1 presents the distribution of demographic characteristics in the case and control groups.
| Variables | Case (N = 60) | Control (N = 60) | Total (N = 120) | P-Value |
|---|---|---|---|---|
| Male gender, n (%) | 40 (66.7) | 40 (66.7) | 80 (66.7) | 1.000 a |
| Alcohol use, n (%) | 4 (6.7) | 7 (11.7) | 11 (9.2) | 0.343 a |
| Cigarette use, n (%) | 24 (40) | 21 (35) | 45 (37.5) | 0.572 a |
| Age, mean ± SD | 58.6 ± 10.5 | 57.4 ± 10.8 | 58.0 ± 10.6 | 0.522 b |
| Age at diagnosis, median (IQR) | 38 (28.3 - 46) | 33.5 (24 - 46.8) | 35 (25 - 46) | 0.105 c |
| HBsAg clearance age, mean ± SD/median (IQR) | 53.2 ± 11.3/53 (47 - 60.8) | - | - | - |
| Body Mass Index, median (IQR) | 27.9 (25.5 - 30.9) | 27.2(24.8 - 28.4) | 27.4 (25.1 - 29.9) | 0.139 c |
| HBsAg clearance period, mean ± SD/median (IQR) | 15.0 ± 7.3/15 (8 - 21) | - | - | - |
| Follow-up period, median (IQR) | 19.5 (13 - 25) | 23 (16 - 25) | 22 (14 - 25) | 0.199 c |
Demographic Characteristics of the Case and Control Group
The presence of comorbidities was comparable between the case and control groups (P > 0.05) (Table 2). Among the seven patients diagnosed with immunosuppressive conditions, one (14.3%) had ankylosing spondylitis, one (14.3%) had multiple sclerosis, one (14.3%) had myasthenia gravis, two (28.6%) had rheumatoid arthritis, and two (28.6%) had undergone renal transplantation.
| Comorbidity | Case (N = 60) | Control (N = 60) | Total (N = 120) | P-Value b |
|---|---|---|---|---|
| Diabetes mellitus | 7 (11.7) | 12 (20) | 19 (15.8) | 0.211 |
| Hypertension | 19 (31.7) | 17 (28.3) | 36 (30) | 0.690 |
| Coronary artery disease | 6 (10) | 2 (3.3) | 8 (6.7) | 0.143 |
| Malignancy | 4 (6.7) | 5 (8.3) | 9 (7.5) | 0.729 |
| Other immunosuppressive diseases | 3 (5) | 4 (6.7) | 7 (5.8) | 0.697 |
Comorbidity Distribution in Case and Control Groups a
There was no statistically significant difference in baseline ALT and HBV DNA levels between the case and control groups (Table 3).
| Parameters | Case (N = 60) | Control (N = 60) | Total (N = 120) | P-Value b |
|---|---|---|---|---|
| Baseline ALT (U/L) | 27 (17.3 - 37.5) | 26 (20 - 43.3) | 26.5 (18 - 39.8) | 0.560 |
| Baseline HBV DNA (IU/mL) | 223 (25 - 3, 120.5) | 1580 (20.25 - 16, 240) | 525 (22 - 5, 552.5) | 0.134 |
Baseline Alanine Aminotransferas and Hepatitis B Virus Deoxyribonucleic Acid Levels: Case vs. Control a
Antiviral therapy was significantly more common in the control group (P < 0.001). The use of interferon (IFN) and nucleos(t)ide analogs (NAs) combined (P = 0.042), as well as NA monotherapy (P < 0.001), was also significantly higher among control group patients. Additionally, the duration of antiviral drug use was longer in the control group (Table 4).
| Parameters | Case (N = 60) | Control (N = 60) | Total (N = 120) | P-Value |
|---|---|---|---|---|
| Antiviral use | 19 (31.7) | 49 (81.7) | 68 (56.7) | < 0.001 b |
| IFN and NA use | 6 (10.0) | 14 (23.3) | 20 (16.7) | 0.042 b |
| NA use only | 13 (21.7) | 34 (56.7) | 47 (39.2) | < 0.001 b |
| IFN use only | - | 1 (1.7) | 1 (0.8) | 0.315 b |
| IFN and NA use period | 9.5 (6.25 - 12.0) | 16.8 (13.3 - 24.1) | 15.0 (10.5 - 21.0) | 0.011 c |
| NA use only period | 5.5 (3.75 - 8.0) | 9.0 (6.0 - 15.0) | 7.0 (5.0 - 13.5) | 0.010 c |
Comparison of Treatment Use Between Case and Control Groups a
4.2. Interferon Gamma-Inducible Protein-10 Polymorphisms
In addition to the c.-135C>T polymorphism identified in the promoter region, two additional polymorphisms — c.85C>T and c.83T>G — were detected in exon 2 of the IP-10 gene. Five patients were excluded from the study due to failure to meet the analytical criteria. Furthermore, in one patient from the case group, the region containing the c.-135C>T polymorphism was deemed suitable for analysis, whereas the regions for the c.85C>T and c.83T>G polymorphisms were not. As a result, only the c.-135C>T polymorphism was assessed in this individual. When the allele frequencies and genotypes of the c.-135C>T, c.85C>T, and c.83T>G polymorphisms were compared between the case and control groups, no statistically significant differences were observed (P > 0.05) (Table 5).
| Variant | Case (N = 57) | Control (N = 58) | Total (N = 115) | P-Value b |
|---|---|---|---|---|
| c.-135C>T genotype/allele | ||||
| CC | 37 (61.7) | 41 (68.3) | 78 (65) | 0.513 |
| CT | 18 (30) | 12 (20) | 30 (25) | 0.513 |
| TT | 2 (3.3) | 4 (6.7) | 6 (5) | 0.513 |
| CA | - | 1 (1.7) | 1 (0.8) | - |
| C alleles | 92 (80.7) | 95 (81,9) | 187 (81.3) | 0.816 |
| T alleles | 22 (19.3) | 20 (17.2) | 42 (18.3) | 0.952 |
| A alleles | - | 1 (0.9) | 1 (0.4) | 0.320 |
| Variant | Case (N = 56) | Control (N = 58) | Total (N = 114) | P-Value b |
| c.85C>T genotype/allele | ||||
| CC | 55 (91.7) | 55 (91.7) | 110 (91.7) | 0.435 |
| CT | 1 (1.7) | 3 (5) | 4 (3.3) | 0.435 |
| C alleles | 111 (99.1) | 113 (97.4) | 224 (98.2) | 0.983 |
| T alleles | 1 (0.9) | 3 (2.6) | 4 (1.75) | 0.322 |
| c.83T>G genotype/allele | ||||
| TT | 50 (83.3) | 54 (90) | 104 (86.7) | 0.543 |
| TG | 6 (10) | 4 (6.7) | 10 (8.3) | 0.543 |
| T alleles | 106 (94.6) | 112 (96.55) | 218 (95.6) | 0.224 |
| G alleles | 6 (5.35) | 4 (3.4) | 10 (4.4) | 0.500 |
Allele and Genotype Frequencies in Case and Control Groups a
5. Discussion
As one of the main causes of cirrhosis, hepatocellular carcinoma, and liver-related mortality globally, chronic HBV is still a significant infectious illness (21). It is evident that the host immune system plays a pivotal role in determining the development of persistence or clearance during the follow-up period of HBV infection (22). One of the most significant chemokines involved in the proinflammatory response is the IP-10 molecule. Several studies have demonstrated a link between serum IP-10 levels and HBsAg loss. For example, Jaroszewicz et al., Wong et al., and Yuan et al. have all reported that IP-10 levels — either elevated or reduced depending on the disease stage and sampling time — are significantly associated with the likelihood of HBsAg clearance (23-25). In our study, we examined the association between HBsAg loss and polymorphisms in the promoter, exon 1, intron 1, and exon 2 regions of the IP-10 gene in patients with chronic hepatitis B infection. While our study did not demonstrate a statistically significant association between the c.-135C>T, c.85C>T, and c.83T>G polymorphisms in the IP-10 gene and HBsAg loss, this finding should not be interpreted as conclusive evidence against a role for IP-10 in HBV immunity. Rather, our results suggest that these specific variants may have limited predictive value within the context of our cohort. The IP-10 is known to play a complex role in antiviral immune regulation, and its expression and function are likely influenced by a broader network of gene-environment interactions, epigenetic mechanisms, and immune signaling pathways. The lack of association observed in our study may reflect the multifactorial nature of HBsAg clearance, which cannot be explained by individual single nucleotide polymorphisms (SNPs) alone.
The rates of HBsAg clearance differ depending on the cohort studied and the inclusion criteria applied. For example, Buechter et al. followed 371 chronic HBV patients for approximately 12 years and reported an HBsAg loss rate of 7.8% (26). Similarly, Habersetzer et al. conducted a six-year follow-up study involving 315 chronic HBV patients in France, observing an HBsAg clearance rate of 9.2% (27). On the other hand, Chien et al. tracked 235 HBeAg-negative chronic HBV patients over seven years and found a lower HBsAg loss rate of 3.4% (28). In our own study, among 1,950 patients monitored from 2005 to 2022, 5.33% experienced HBsAg clearance. Consistent with previous research, these differences are likely influenced by multiple factors such as patient demographics, ethnic background, disease stage, duration of follow-up, and use of antiviral therapies. Studies have shown that HBsAg clearance is associated with older age and longer follow-up. Chu and Liaw demonstrated that clearance rates rise over time (6), and Buechter et al. reported a mean seroconversion period of 12 years (26). In line with these findings, our study observed a median HBsAg loss time of 15 years, with most cases occurring after 10 years. We also found that clearance was more frequent in individuals over 40. A large cohort study and a separate study from South Korea both reported higher rates of HBsAg seroclearance in patients above 40 compared to younger individuals (29, 30). In our cohort, the average age at the time of HBsAg loss was 53.2 years, and 85% of the cases occurred in patients older than 40. These results highlight age and follow-up duration as important factors in predicting HBsAg clearance.
Prior investigations suggest that certain SNPs in the IP-10 gene promoter are linked to the severity of infectious diseases like pulmonary tuberculosis and cerebral malaria (17). The c.-135C>T (also referred to as G-201A in earlier genome builds) variant has been shown in multiple studies to influence IP-10 expression levels, supported by findings from GWAS (31), protein QTL (32), and its association with HBV and HCV infections. While Talaat et al. reported no correlation between IP-10 polymorphisms and disease progression in chronic HCV patients, Thanapirom et al. highlighted the importance of the -135G/A polymorphism in achieving a sustained virologic response to pegylated IFN therapy (17, 18). The A allele of the G-201A polymorphism in the promoter region of the IP-10 gene has been associated with increased disease severity and progression. In a large-scale study conducted in China involving 2,400 individuals with chronic HBV infection, Deng et al. reported a higher frequency of disease progression among male carriers of the A allele (19). Similarly, Xu et al. found that this polymorphism was linked to liver disease development in HBV-infected patients, with the A allele being more prevalent in those with advanced disease (20). A separate study from Thailand demonstrated an association between the G-201A variant and both reduced HBsAg levels and improved virologic response to pegylated IFN therapy. Patients carrying the GG genotype exhibited lower HBsAg levels and higher response rates; however, no significant difference in HBsAg clearance rates was observed between GG and non-GG genotypes (16). Collectively, these studies suggest that the c.-135C>T polymorphism has previously been examined in the context of disease progression in HCV and HBV, as well as in relation to HBsAg level changes during IFN-based therapy. In contrast, our study directly investigated the association between this polymorphism and HBsAg clearance, finding no statistically significant correlation. Interestingly, we also identified a previously unreported CA genotype in one individual from the control group. Further genetic and functional analyses are needed to determine the clinical significance and potential biological implications of this novel variant. Although c.85C>T has not been linked to HBV, its potential role has been investigated in GWAS and protein QTL studies (31, 33). A study investigating the influence of genetic and lifestyle factors on circulating biomarkers related to inflammation and cancer identified the c.85C>T polymorphism in the IP-10 gene (34). In our research, neither the c.85C>T nor the c.83T>G polymorphism showed a significant association with HBsAg loss. The c.83T>G variant is a novel variant not reported in gnomAD v4.1, TOPMed, or the Turkish Variome, and we considered it potentially significant based on our observations. Therefore, we believe that our study is the first to evaluate the potential association of the c.-135C>T, c.85C>T, and c.83T>G polymorphisms with HBsAg loss in patients with chronic HBV infection. Given the absence of significant associations between the studied IP-10 polymorphisms and HBsAg loss, several methodological and biological factors should be considered. One key limitation is the relatively small sample size, which may have limited the statistical power to detect associations, particularly for low-frequency variants. The SNPs with low minor allele frequencies often require larger cohorts to reach adequate power, especially when investigating complex traits such as viral clearance. Additionally, population-specific genetic backgrounds may have influenced our results.
For instance, the allele frequency of the c.-135C>T polymorphism is reported as 0.402% in the Turkish population according to gnomAD v4.1, whereas it was 18.3% in our cohort. Similarly, the c.85C>T variant has a reported allele frequency of 10.737% in gnomAD, compared to 1.75% in our study population. These discrepancies likely reflect differences between the general population and disease-specific cohorts and may also point to underlying ethnic substructure within the Turkish population (35, 36). Moreover, previous studies that reported associations between IP-10 polymorphisms and HBV-related outcomes were primarily conducted in East Asian populations, where allele distributions and linkage disequilibrium patterns differ substantially from those in our cohort (19, 20). Therefore, population-specific factors must be taken into account when interpreting genetic association studies across diverse ethnic backgrounds. Beyond sample size and population variability, it is also important to consider the biological complexity of HBsAg clearance when interpreting these findings. The investigated polymorphisms may exert only a modest or indirect influence on IP-10 function or expression. HBsAg clearance is a multifactorial event involving a complex interplay between host genetics, immune response, viral factors, and treatment exposure. It is plausible that these individual variants alone are not sufficient to predict seroclearance without considering gene-gene and gene-environment interactions (37).
In addition to genetic and population-specific factors, treatment-related variables may have influenced our findings. Although we observed a significantly lower frequency and shorter duration of antiviral therapy among patients with HBsAg loss, treatment effects were not fully controlled for in the analysis. Given that antiviral therapy is a major modulator of immune-mediated viral clearance, such differences in treatment exposure may have confounded the genetic associations under investigation (38). Another important limitation of our study is the lack of functional analysis to support the observed genetic findings. Our investigation focused solely on genotyping, without evaluating the potential functional consequences of the identified polymorphisms — such as their effects on IP-10 mRNA or protein expression. However, functional validation through transcriptomic, proteomic, or promoter activity assays is essential to establish a mechanistic link between genotype and phenotype. In the absence of such data, the biological relevance of these variants remains speculative (20). Furthermore, the identification of a previously unreported genotype (CA at c.-135) and a novel SNP (c.83T>G) indicates additional genetic diversity within the IP-10 locus that has not been functionally characterized. These rare or population-specific variants may exert unique immunological effects that warrant further investigation in larger, well-characterized cohorts.
Overall, this study provides valuable negative data by demonstrating that the investigated polymorphisms are not reliable predictive markers for clinical outcomes in chronic hepatitis B. The lack of a clear association underscores the complex interplay of host genetic factors in immune-mediated viral clearance, which likely involves multiple genes and signaling pathways beyond IP-10. These findings further highlight the importance of investigating other genetic and immunological determinants — such as cytokine gene polymorphisms, HLA alleles, and components of the innate immune response — to better elucidate the mechanisms driving both spontaneous and treatment-induced HBsAg loss.
5.1. Conclusions
In conclusion, our findings indicate that the c.-135C>T, c.85C>T, and c.83T>G polymorphisms in the IP-10 gene are not significantly associated with HBsAg loss in patients with chronic hepatitis B. However, this result should not be interpreted as definitive evidence against the involvement of IP-10 in HBV-related immune responses. Given the complexity of HBsAg clearance, these specific SNPs may have limited predictive value in isolation. Moreover, the identification of a novel variant (c.83T>G) and an unreported genotype (CA at c.-135) suggests underlying genetic diversity within the IP-10 locus that warrants further exploration. To validate these findings and explore potential population-specific effects, future studies with larger and more ethnically diverse cohorts are needed. Furthermore, in-depth investigations should be conducted to assess the combined effects of IP-10 expression levels and promoter region polymorphisms on HBsAg clearance and treatment response. As a future direction, we also recommend integrating functional assays and epigenomic analyses, such as DNA methylation profiling of the IP-10 promoter region, to better understand the regulatory mechanisms that may influence HBsAg loss. Additionally, employing omics approaches, such as transcriptomics and proteomics, to comprehensively investigate the effects of the IP-10 gene may contribute to a deeper understanding of the biological pathways influencing HBsAg loss.
