






1. Background
Hepatitis B virus (HBV) infection has always been an important public health problem in China. Although many infants born to mothers with hepatitis B received a three-dose vaccine at 0, one, and six months, a few children were still infected with HBV. Vaccination failure led to vertical transmission of HBV.
Recent studies reported that mutations in HBV S region were thought to be closely related to the HBV vaccination failure (1-6). Furthermore, HBV surface antigen (HBsAg) is a major binding site for neutralizing antibodies and HBV (2, 7). Amino acid change caused by mutations in the S region can lead to changes in the structure of HBsAg (8). With the change in the structure of HBsAg, the antigenicity of HBsAg and the binding capacity of antibodies with HBsAg decreased. The Major Hydrophilic Region (MHR) is located in aa99 - 169 of HBsAg (9), and contains several B-cell epitopes. Mutations in MHR, especially in “a” determinant (aa124 - 147), were observed in children, who failed vaccination in a previous study (2, 9-11). Among the mutations in MHR, some of them were crucial for the antigenicity of HBsAg (12, 13). These mutations led to immune escape of HBV.
In previous studies, the characteristics of vertical transmission of HBV were analyzed for the consistency of the sequence (14, 15). When the sequence similarity between the mother and child was high, it was considered that the HBV was conserved in the vertical transmission. However, the existence of HBV in the host is in the form of quasi-species and genetically distinct viral species coexist in infected individuals (16). Deep sequencing has a higher sensitivity than sanger sequencing (17), and low-frequency mutations can be detected. Deep sequencing can provide more reliable sequencing data.
2. Objectives
Vertical transmission is the most important route of transmission. Escape mutations in HBV S gene play an important role in vertical transmission due to vaccination failure. In this study, deep-sequencing was used to obtain HBV S gene sequences of mothers and children. From the sequencing data, the characteristics of HBV in vertical transmission due to vaccination failure was described clearly, and showed the influence of low-frequency mutations on vaccination failure.
3. Methods
A total of nine mother-child pairs with chronic HBV were recruited in this study. All children received three doses of vaccine at 0, one, and six months, and were diagnosed as chronic HBV carriers. All children’s mothers were diagnosed as chronic HBV carriers before pregnancy. The serum samples for the mothers and children were collected simultaneously at Children’s Hospital of Chongqing Medical University, China. None of them had received antiviral treatment. This study was approved by the ethical committee of the Children’s Hospital of Chongqing Medical University, and written informed consent was obtained from all adult patients or guardians on behalf of the children enrolled in this study.
Serological markers of HBV (HBsAg, anti-HBs, HBeAg, anti-HBc, anti-HBe) were tested using enzyme linked immunosorbent assay (ELISA) kits (Abbot, USA). Serum HBV DNA load was measured using polymerase chain reaction (PCR) assay kits (Roche, Switzerland). Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were tested using auto biochemical analyzer (Beckman CX9 ALX, USA). They were all measured by clinical laboratory Children’s Hospital of Chongqing Medical University.
The HBV DNA was extracted from 200 µL of serum using a QIAamp ultrasens virus kit (QIAGEN GmbH, Hilden, Germany). The HBV S gene was amplified with PCR and the primer sequences were designed according to one research from China (11). The first amplification primers were P1: 5-GGGTCACCATATTCTTGGGAAC-3 (nt 2814-2835) and P2: 5-GGGGGTTGCGTCAGCAAACAC-3(nt1180 - 1200); The second amplification primers were P3: 5-ACTTTCCTGCTGGTGGCTCC-3 (nt51 - 70) and P4: 5-CATATCCCATGAAGTTAAGG -3 (nt 867 - 886). The PCR was performed using Premix PrimeSTAR HS DNA polymerase (TAKARA, Japan) with the following PCR. The two amplification steps were performed according to the manufacturer’s protocol: 98°C for 10 minutes, followed by 30 cycles of denaturation at 98°C for 10 seconds, annealing at 55°C for 15 seconds, 72°C for 45 seconds, and a final extension at 72°C for 10 minutes. The PCR products were confirmed by electrophoresis on a 2% agarose gel. After imaging analysis on UV transilluminator, all positive PCR products were purified with DNA purification kit (TAKARA, Japan).
Deep sequencing (Genome Analyzer: Illumina HiSeq2500, Beijing, China) was used to detect the HBV S region gene sequences. Firstly, a library of PCR products of the HBV S genome was prepared using the Nextera™ DNA sample prep kit (Illumina, USA). The PCR products were disrupted to 250 to 500 bp of fragments, according to the manufacturer’s protocols. Then, barcode primer was added to 5’-end of the fragments by PCR. Finally, the fragments purified by gel extraction kit were sequenced with HiSeq2500 (MyGenostics, Beijing, China). The detailed experiment of deep sequencing was performed by MyGenostics.
To analyze the sequencing data, the online tool fastqc (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) was used to evaluate the quality of raw data, then removed the reads with quality score below 20 and polyN sequences. Finally, HBV S region gene mapping the assembled sequence reads was mapped based on reference HBV S gene (GenBank accession numbers: AB205119).
The HBV complete genome was downloaded from NCBI, and the genotype of the downloaded HBV complete genome was analyzed using REGA web server (http://newbioafrica.mrc.ac.za/rega-genotype/) to obtain exact genotype sequences. Then, the exact genotype sequences were used to build HBV S gene reference and position specific matrix (PSSM). According to PSSM, the researchers assessed which genotype the reads belonged to.
The PSIPRED was used to predict the secondary structure and the transmembrane regions of HBV surface protein (18). The online tool DISULFIND was used to predict the disulfide bond of “a” determinant (19).
The diversity of the HBsAg amino acids was assessed using the Shannon entropy (SE). It was calculated according to the type of amino acid. The formula is as follows:
Where n is the number of amino acids in one site, and fi is the frequency of various amino acids at that site.
4. Results
4.1. Characteristics of Mother-Child Pairs and Sequencing Data
Table 1 lists the characteristics of nine mother-child pairs. All of the nine mother-child pairs were positive for HBsAg and HBcAb. Seven mother-child pairs were HBeAg-positive, two mother-child pairs were HBeAg-negative in mothers, and their children were HBeAg-positive. The viral load of eight mother-child pairs were > 6 log10 IU/mL, and the viral load of mother in the pair four was < 3 log10 IU/mL. All of the mother-child pairs exhibited normal values or mild liver damage in the liver biochemical tests.
| Patients | Gender | Age, y | HBsAg | HBsAb | HBeAg | HBeAb | HBcAb | ALT, U/L | AST, U/L | HBV DNA, IU/mL | Dominant Genotype |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1C | Male | 8 | + | - | + | - | + | 11.4 | 24.2 | 9.91 × 107 | B |
| 1M | Female | 35 | + | - | + | - | + | 31.0 | 30.0 | 1.19 × 108 | B |
| 2C | Female | 11 | + | - | + | - | + | 29.4 | 28.8 | 1.65 × 106 | B |
| 2M | Female | 36 | + | - | + | - | + | 23.0 | 24.0 | 3.81 × 107 | B |
| 3C | Male | 15 | + | - | + | - | + | 19.0 | 29.0 | 6.21 × 107 | B |
| 3M | Female | 40 | + | - | - | + | + | 7.0 | 25.0 | 2.62 × 108 | B |
| 4C | Male | 7 | + | - | + | - | + | 25.0 | 30.0 | 1.08 × 108 | B |
| 4M | Female | 40 | + | - | - | + | + | 16.0 | 19.0 | < 1.00 × 103 | B |
| 5C | Female | 1.5 | + | - | + | - | + | 38.0 | 42.7 | 1.49 × 106 | B |
| 5M | Female | 27 | + | - | + | - | + | 11.0 | 17.0 | 5.83 × 107 | B |
| 6C | Female | 3 | + | - | + | - | + | 20.0 | 40.0 | 4.14 × 107 | C |
| 6M | Female | 30 | + | - | + | - | + | 49.0 | 38.0 | 3.97 × 107 | C |
| 7C | Female | 0.8 | + | - | + | - | + | 23.3 | 32.3 | 2.34 × 107 | C |
| 7M | Female | 27 | + | - | + | - | + | 23.0 | 23.0 | 8.99 × 107 | C |
| 8C | Female | 4 | + | - | + | - | + | 42.0 | 44.0 | 3.83 × 107 | B |
| 8M | Female | 30 | + | - | + | - | + | 37.0 | 35.0 | 1.99 × 108 | B |
| 9C | Male | 7 | + | - | + | - | + | 44.0 | 44.0 | 1.16E + 07 | B |
| 9M | Female | 32 | + | - | + | - | + | 9.0 | 20.0 | 2.76E + 07 | B |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate amino transferase; C, child; HBV, hepatitis B virus; HBsAg, hepatitis B surface antigen; HBsAb, hepatitis B surface antibody; HBcAb, antibody to hepatitis B core antigen; HBeAg, hepatitis B e antigen; M, mother.
After all samples were sequenced with deep sequencing, a total of 4,452,107 data were obtained. Filtering out low-quality and polyN data, 4,145,246 high-quality data were obtained. Then these data were sorted according to barcode sequences and data that could not be categorized were filtered; 1,730,005 sequencing data was finally obtained. The data utilization was 38.86%. The sequencing data of all the samples could completely cover the HBV S region gene with a coverage rate of 100%.
4.2. High Conservative of HBV S Region in Vertical Transmission
The analysis results showed that genotypes of nine mother-child pairs were all B+C mixed genotypes. The dominant genotype was B in 7/9 pairs of mother-child, and C in 2/9 pairs (Table 1). The dominant genotype of mothers was the same as that of children. In other words, the dominant genotype did not change during vertical transmission.
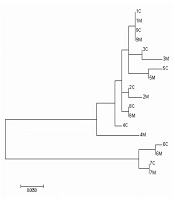
A phylogenetic tree (Figure 1) was constructed by maximum likelihood algorithm using consensus sequences. Sequences from the same genotype were clustered together. Overall, 8/9 of the mother-child pairs were clustered together except pair four. Though pair four was not clustered together, the identity is also higher than 98.5%.

Maximum likelihood phylogenetic tree based on HBV S consensus sequences from 9 pairs of mother-child pairs. M, mother; C, child.
Consistency analysis can only compare the difference between the two DNA strands in the mother and child, yet HBV DNA was diverse in the host, so that the comparison of diversity between mothers and children was also necessary. The Shannon entropy was used to assess the diversity of amino acids. By comparing the mean Shannon entropy for the same site of amino acids (Figure 2), the researchers found that the diversity of most amino acid sites had a small difference between mothers and children (t-test, P > 0.05). The trend of diversity is basically consistent between mothers and children.

The amino acids diversity of mothers and children in HBV S region gene. The X-axis denotes the S amino acid site and the Y-axis denotes the entropy of the sites.
After comparing the diversity, the researchers analyzed the mutation sites of each mother-child pair; it was found that the mutations shared by mothers and children made up a high proportion of children’s mutations and the rate was 83.13% ± 8.45%. The mutations of children most came from their mothers.
The above results indicated that the mutations were remarkably consistent and HBV S gene was highly conserved in vertical transmission.
4.3. The Influence of Mutations in HBsAg Region
To investigate the relationship between mutations and vaccine failure, the S gene sequences (from Genbank) of 27 mothers served as controls. The sequences were from one research in Thailand (20) and all mothers were HBsAg-positive and their infants were not infected with HBV and received a complete course of hepatitis B vaccination. The accession numbers of S gene sequences are shown in Supplementary File Appendix 1.
In the current study, many point mutations that can cause amino acid change were found in each mother-child pair by analyzing the sequencing data (Table 2). The mutations were distributed throughout S gene. To explore the impact of mutations, the researchers analyzed the S region gene sequences of mothers and children. In MHR, sG102D sP108L sL109I sI110L/V sG112R sS117G sG119R sP120Q/R/L sT123I were observed in upstream (aa99 - 123) of “a” determinant, and sY/F161C sL162V were observed in downstream (aa148 - 169) of “a” determinant. The mutation rate was 29.67% ± 5.09% in upstream and 9.85% ± 0.03% in downstream. The mutation rate was higher in upstream of “a” determinant. In “a” determinant, point mutations were found in aa 124 126 133 137 139 140 141 142 145. From Table 2, the mutated position was more frequent in second loop of “a” determinant than that in the first loop. The mutation rate was 33.33% ± 9.07% in second loop and 6.75% ± 4.44% in first loop. The frequency of the mutations, including sC124G/S sT126I sM133L sT143S, were low in nine mother-child pairs, and the following mutations were frequent in nine mother-child pairs, including sK141R sC137S sT140I sK141R sG145R sC139Y sK141R sP142L. From Table 2, it was found that sC137S sC139Y sT140I sK141R sP142L sG145R occurred in both mothers and children in some mother-child pairs. In the above-mentioned immune mutations, sG145R sT143S and sT126I were proved to be associated with immune escape. In the current study, sC137S or sC139Y, which were located in key position of “a” determinant and rarely reported in previous studies, were found in all mother-child pairs, and sC137S or sC139Y were also observed to occur in both mothers and children.
| 102G | 107C | 108P | 109L | 110I | 112G | 113S | 117S | 119G | 120P | 123T | 124C | 126T | 133M | 137C | 139C | 140T | 141K | 142P | 143T | 145G | 160K | 161Y/F | 162L | 164E | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1M | S | L | I | L/V | R | T/G | G | I/N | S | I | R | R | R | C | |||||||||||
| 1C | S | L | I | L/V | R | T | G | S | I | R | R | R | C | ||||||||||||
| 2M | S | L | I | L/V | R | T | L | S | I | R | R | R | C | ||||||||||||
| 2C | S | L/H | I | L/V | R | T | R | Q/R/L | S | I | R | R | R | C | V | G | |||||||||
| 3M | D | S | L | I | L/V | R | T | G | R | Q/R | I/N | G/S | S | I | R | R | R | C | |||||||
| 3C | S | L | I | L/V | R | T | G | I/N | S | I | R | L | R | R | C | V | |||||||||
| 4M | D | S | L | I | L/V | R | T | G | I/N | S | I | R | R | R | C | ||||||||||
| 4C | S | L | I | L/V | R | T | G | R | I/N | I | S | I | R | S | R | R/I | C | ||||||||
| 5M | S | L | L/V | R | T | I/N | S | I | R | R | R | C | V | ||||||||||||
| 5C | S | L | I | L/V | R | T | G | S | I | R | R | R | C | ||||||||||||
| 6M | D | L | I | R | R | Q/R | Y | R | L | V | |||||||||||||||
| 6C | D | S | L | I | R | R | Q/R | I | Y | R | L | C | V | ||||||||||||
| 7M | D | L | I | R | G | R | Q/R | Y | R | L | V | ||||||||||||||
| 7C | D | S | L | I | R | G | R | Q/R | S | Y | R | L | R | C | V | ||||||||||
| 8M | S | L | I | L/V | R | T | G | Y | R | L | R | C | V | ||||||||||||
| 8C | S | L | I | L/V | R | T | G | Y | R | L | R | V | |||||||||||||
| 9M | S | L | I | L/V | R | T | G | S | I | R | R | R | C | ||||||||||||
| 9C | S | L | I | L/V | R | T | S | R | C |
Abbreviations: C, child; M, mother.
In the current study, a high frequency of variants in the viral population occurred in the major hydrophilic region. Many of them were delivered to the offspring by their mothers. Among them, the mutations sC137S or sC139Y that could lead to change of “a” determinant structure were observed in all mother-child pairs.
4.4. Secondary Structure Analysis of HBV Surface Protein
The secondary structure of “a” determinant is presented as a double loop, formed through disulfide bonds between cysteine residues 124 and 137 and residues 139 and 147 (10, 21, 22). The double loop structure had an important influence on HBsAg immunogenicity and antigenicity. If the loop structure was damaged due to the point mutations, the affinity of anti-HBs was decreased. In the current study, sC137S sC139Y were observed in “a” determinant in all mother-child pairs. To explore the effect of sC137S or sC139Y on the secondary structure of HBV surface protein, the secondary structure was predicted using PSIPRED and the confidence of disulfide bonding state prediction was predicted using DISULFIND.
As shown in Figure 3, compared with the secondary structure of the wild-type HBV surface protein, the quantity and location of the transmembrane structure of sC137S and sC139Y did not change. However, the disulfide bonds of the “a” determinant were predicted and it was found that when one of the sC137S or sC139Y occurred, the disulfide bond in both loop structures would be affected. A mutation in one loop reduced the likelihood of another loop disulfide bond formation.

Predicted transmembrane domains of HBsAg and confidence of disulfide bonding state prediction. A, The predicted transmembrane domains of HBsAg for WT, C137S and C139Y; WT, wild type. B, confidence of disulfide bonding state prediction of “a” determinant for WT, C137S and C139Y; the mutations were in red frame. C, Model of the “a” determinant in HBsAg, circles represent amino acids and solid lines were of disulfide bonds. D, model of the “a” determinant in HBsAg with mutations, circles represent amino acids and solid lines were of disulfide bonds, red lines represent that the disulfide bonds were broken.
5. Discussion
In the current study, most of the mother-child pairs were HBeAg-positive. In a previous study, it was proved that HBeAg could inhibit immune response of the host. The mutation frequency of HBV DNA was low when the host maintained HBeAg-positivity in the long term (23).
Previous studies showed that HBV was highly conserved in vertical transmission (14, 15, 23). However, most of them only compared the identity of DNA sequences between mothers and children. The results coincided with the current study. However, HBV existed in host in the form of quasi-species (16), and the HBV DNA strains were diverse. Analyzing the HBV DNA sequences consistency alone is not comprehensive enough, and it is necessary to analyze the DNA diversity of HBV.
In this study, the deep sequencing was used to detect the mutations in the HBV S region. Through analyzing the sequencing data, some relatively low frequency of mutations was found in all mother-child pairs. The sequence diversity, identity, and mutations of HBV S region indicated that HBV in the host maintains a fairly stable state. All of these indicated that HBV S gene maintained high consistency and had high conservation in vertical transmission. High conservative vertical transmission suggested that the immune escape mutations that pre-existed in mothers were transmitted to the offspring through vertical transmission and led to vaccination failure.
Previous studies have confirmed that the failure of HBV is closely related to the immune escape mutation of S gene, especially in the “a” determinant located in MHR (1, 3, 7, 9, 24, 25). In the current study, all mothers and children were both observed to have mutations in major hydrophilic region or “a” determinant. In another study, it was found that immune escape mutations existed in mothers of infants with immunoprophylaxis failure by deep sequencing (26). Due to the immune escape mutations that preexisted in mothers and transmitted to children, vaccination failure occurred.
The “a” determinant exists in all genotypes and is the main target of antibodies (10). Antibodies against the “a” determinant can provide immune protection to all genotypes of HBV. The double-loop structure of “a” determinant is formed through the disulfide bonds between four cysteine, which are located in aa124 and aa137, aa139 and aa147 (10, 21, 22). In a previous study, the researcher constructed cyclical and linear structure of “a” determinant and compared the antibody affinities of the two peptides (21, 22). The result showed that the antibody affinity of cyclical peptide was significantly higher than linear peptide. In this study, the mutations in aa137 or aa139 were found in almost all mothers and children. Cysteine was mutated in serine and tyrosine in aa137 and aa139. When mutations occurred in aa137 or aa139, the disulfide bonds were damaged and the cyclic structure became a linear structure. The affinity of antibody against HBsAg decreased significantly. From the results of disulfide bond analysis, the researchers found that sC137S or sC139Y in one loop reduced the likelihood of another loop disulfide bond formation. The changed secondary structure decreased the immunogenicity and led to immune escape.
Except for the above two mutations, many mutations were also found in “a” determinant in the current study. The sG145R mutation was the most commonly detected immune escape variant, which could reduce the affinity of antibody for the HBsAg (1, 7). The 3D structure of MHR constructed by former researchers showed that aa126 and aa143 were located on the surface of HBsAg (27, 28). In this study, sT143S was only found in one child; previous studies indicated that sT143S single substitution had a slight alteration in antigenicity, at aa143 which was located in second loop of “a” determinant. The sT143S can be observed in various genotypes of HBV. Compared with sT143S, sT126I which was located in the first loop of the “a” determinant of HBsAg was mainly found in genotype C and can result in significant decrease in the antigenicity. Furthermore, sT126I can change chemical properties and reduce the antigenicity due to the structural changes of the HBsAg. The following three mutations, sK141R sP142L sT140I, were rarely observed in studies about immune escape. However, in the current study, these three mutations nearly occurred in nine pairs of mother-child pairs. Their role in immune escape is unknown, and all of them were located in the second loop of “a” determinant.
Outside “a” determinant yet in MHR, many mutations were also found, including sG102D sC107S sP108L sL109I sI110L/V sG112R sS113T/G sS117G sG119R sP120Q/R/L sT123I sK160R sY161F/C sL162V sE164G. A previous study showed that the region between aa120 and aa123 was vital to antigenicity of HBsAg (29). The mutation occurred in aa120 seriously affected the affinity of HBsAg to anti-HB antibodies, thus indicating that this mutation was related to immune escape. The mutation that occurred in aa123 was mainly associated with impaired secretion of HBsAg and led to occult HBV infection. sI110L sF161Y had been found in a previous study on immune escape mutations (30-32). However, both two mutations were only found in genotype C in the current study, and sI110L was only observed in genotype B in a previous study, whether it was crucial for immune escape, was unknown in genotype B. The rest of mutations were rarely reported in the past, yet were observed in almost all mothers and children in the current study. It was unclear whether they could influence immune escape.
5.1. Conclusions
In conclusion, the HBV S gene was highly conservative in vertical transmission when the host maintained immunosuppression in the long-term. High conservative vertical transmission suggested that the immune escape mutations were transmitted to the offspring through vertical transmission. Since immune escape mutations already existed in children before vaccination, the vaccine could not prevent HBV infection with the host.
