







1. Background
It is estimated that at least 71 million people are living with chronic HCV infection and are at risk for liver diseases such as cirrhosis and hepatocellular carcinoma. Approximately 1.8 million individuals are newly infected annually (1, 2). Although direct-acting antivirals (DAAs) have considerably improved the treatment of patients with chronic HCV infection, several limitations of this management underscore the need for the development of a prophylactic vaccine to control HCV infection worldwide. The limitations include high cost of treatment, risk of reinfection, progress of liver disease despite cure of HCV infection, DAA resistant HCV variants and need to implementation of complex and expensive screening programs to identify HCV infected individuals as only 5% of HCV cases worldwide are aware of their infection (3).
There is increasing evidence that an effective HCV vaccine requires not only potent T-cell responses but also high levels of neutralizing antibodies (NAbs) (4). The role of broadly NAbs in the spontaneous clearance of HCV in cute infection and also against the progression of liver disease in chronic HCV infection has been well-known (5). At present, one of the two main vaccine strategies which has found its way to the human trials is the use of a recombinant form of HCV envelope glycoproteins gpE1/gpE2 (6). This strategy in addition to the production of broadly NAbs that neutralize HCV infectivity activates IFN-γ producing T cells, which facilitate viral clearance. However, the results obtained from the strategies developed to elicit antibodies against HCV show that further work and optimization are needed to identify the ideal vaccine antigens and adjuvants, as well as an effective vaccine delivery platform.
A variety of platforms have been used to introduce viral antigens in an immunogenic way. Among them, vaccination with DNA plasmids that express endogenous antigen from non-replicating and non-transmissible genetic materials provides a safe and reliable platform for the development of next-generation vaccines (7). DNA vaccines induce both cellular and humoral immune responses (7). Several prior reports indicated the application of DNA vaccines for the induction of immune responses against HCV antigens and epitopes (8-10). However, DNA vaccines with relatively weak immunogenicity require improvement. For this purpose, the formation of virus-like particles by the fusion of HBsAg (9-11) or the inclusion of universal helper epitopes and endoplasmic reticulum signal sequences (8) was suggested. Among various strategies have been used for this intent, the use of genetic adjuvants is a promising approach (12). It has been shown that heat-shock proteins (HSPs) act as potent adjuvants. In this regard, Gp96 protein as a member of HSPs family and/or its N-terminal domain have been used in some experiments as an adjuvant (13).
2. Objectives
In this study, we designed and constructed a DNA vaccine (pcDNA-E2-NT(gp96)) that encodes a fusion protein composed of HCV E2 ectodomain and N-terminal domain of gp96 as adjuvant, and evaluated its potential immunogenicity as an HCV candidate vaccine in BALB/c mice.
3. Methods
3.1. In silico Design, Modeling and Validation of Fusion Protein
To generate a fusion protein composed of HCV E2 ectodomain (E2) and N-terminal domain of gp96 (NT(gp96)), the E2 ectodomain sequence (aa 384-661, genotype 1a, Gen Bank accession No.: AF011753.1) and gp96 sequence from Xenopus laevis (GenBank accession number AY187545.1) were extracted from NCBI database (https://www.ncbi.nlm.nih.gov). The N-terminal domain sequence of gp96 was fused to N- or C-terminal of E2 ectodomain sequence with flexible linker (GSGGGG) using BioEdit Sequence Alignment Editor (http://www.mbio.ncsu.edu/BioEdit/bioedit.html), and two possible fusion forms, including NT(gp96)-E2 and E2-NT(gp96) were produced. The secondary and tertiary structures of fusion proteins were predicted using GOR4 (https://npsa-prabi.ibcp.fr/cgi-bin/npsa) (14) and I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) server (15), respectively. The quality of the predicted three dimensional (3D) models was analyzed and validated using protein structure analysis (ProSa) server (https://prosa.services.came.sbg.ac.at/prosa.php) (16).
3.2. Protein-Protein Interaction (Docking) Study
In order to evaluate the interaction of fusion protein (E2-NT(gp96)) with the primary receptor of HCV (CD81), protein-protein docking was performed using Hex server (http://www.loria.fr/~ritchied/hex/). Hex is an interactive protein docking program, which identifies pairs of molecules only using their 3D knowledge. Hence, 3D shapes of the E2-NT(gp96) modeled by the I-TASSER and the CD81 extracted from the Protein Data Bank (http://www.rcsb.org/pdb/) were subjected to the server.
3.3. In vitro Expression Assay
For in vitro expression assay, the E2-NT(gp96) fragment was subcloned into pEGFP-N3 vector and its expression was evaluated in COS-7 cells. To generate the pEGFP-E2-NT(gp96), the E2-Linker gene was subcloned from the synthesized construct (pBluescript-E2-Linker, synthesized by Biomatick company, Canada) into the Nhe I and EcoRI sites of pEGFP-N3 plasmid (Clontech, USA). Subsequently, the gene fragment encoding for the N-terminal domain of gp96 (NT(gp96)) was excised by EcoRI and Apa I restriction enzymes from pQE-30-gp96 vector (17), and subcloned into corresponding sites of the pEGFP-E2-Linker generating pEGFP-E2-NT(gp96). Constructed vectors were confirmed by restriction analysis and DNA sequencing reactions. To demonstrate in vitro expression of pEGFP-E2-NT(gp96), up to 5 × 104 COS-7 cells per well of a four-well plate (Greiner, Germany) were cultured in Dulbecco’s Modified Eagle’s medium (DMEM, Gibco) and incubated. In vitro expression of pEGFP-E2-NT(gp96) was evaluated by transfection of these cells with 5 µg of pEGFP-E2-NT(gp96), pEGFP-N3 (positive control) and pcDNA3.1 (negative control) plasmids in separate reactions using LINPEI 25KDa (10 µM; poly sciences, Europe) as described previously (13). The expression of the proteins was confirmed by observation of the EGFP signal under a fluorescence microscope (Nikon E200, USA) at 24 hours post-transfection.
3.4. Construction and Preparation of pcDNA-E2-NT(gp96) Plasmid
To construct recombinant pcDNA-E2-NT(gp96), E2-Linker and NT(gp96) fragments were cloned into pcDNA3.1(+) (Invitrogen, Carlsbad, CA). The E2-Linker DNA subcloned from pBluescript-E2-Linker vector into the Nhe I and Not I restriction sites of pcDNA3.1(+) generating pcDNA3.1-E2-Linker. The N-terminal segment of gp96 was amplified from pQE-30-gp96 plasmid (17) using a forward primer (5-ATATGCGGCCGCGAAGATGACGTTG-3) harboring Not I restriction site and a reverse primer (5-GGGCTCTCTAGATTATTTGTAGAAGGCTTTG-3) harboring Xba I restriction site. Amplified DNA was cloned into the recombinant pcDNA3.1-E2-Linker plasmid between Not I and Xba I restriction sites generating pcDNA3.1-E2-Linker-NT(gp96). Constructed plasmid named pcDNA-E2-NT(gp96). The pcDNA-E2-NT(gp96) plasmid was purified by ion-exchange chromatography with Endo Free plasmid Giga Kit (QIAGEN, Valencia, CA). The precision and accuracy of the construct were confirmed by restriction analyzing and DNA sequencing.
3.5. Mice Immunization
Specific-pathogen-free female BALB/c mice (6 - 8 weeks old) were purchased from Laboratory Animals Centre, Pasteur Institute of Iran, and maintained at the Animal Holding Unit of the Tehran University of Medical Sciences. Eight mice per group were immunized intramuscularly with a total volume of 100 µL (50 µL per thigh) of antigen or control formulation at week 0, 3 and 6. The mice were received 50 µg of the pcDNA-E2-NT(gp96) plasmid encoding the fusion protein E2-NT(gp96) or pcDNA3.1(+). As a negative control, a group of mice (n = 8) was immunized with 100 µL phosphate-buffered saline (PBS, pH 7.3). Serum samples were collected before immunization and 2 weeks after the last injection. All animals received humane care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86 - 23 revised 1985), (code of ethics; IR.TUMS.VCR.REC.1395.177).
3.6. Enzyme-Linked Immunosorbent Assay (ELISA)
The level of E2-specific serum IgG and isotypes (IgG1 and IgG2a) of the immunized mice were determined by ELISA (13). The ratio of IgG2a/IgG1 isotype responses was also determined. In brief, ninety-six-well microtiter plates (Costar 3690) were coated with 100 µL (1 µg/mL in PBS) of recombinant E2 protein at 4°C overnight. The wells were washed four times with PBST (PBS, 0.05% Tween 20), and blocked with 5% skim milk in PBS for 2 hours at 37°C. After three times washing, 100 µL of sera diluted 1:500 in dilution buffer (PBST, 1% skim milk) was added to the plate in duplicates and incubated for 2 hours at 37°C. The wells were washed four times, and 100 µL of the HRP-conjugated goat anti-mouse IgG, IgG1 or IgG2a (1:3000 dilutions, Southern Biotech, Birmingham, AL) was added and incubated for 2 hours at 37°C. After four times more washing, the color was developed by adding 100 µLTMB substrate (3,3-5,5-tetramethylbenzidine, Sigma) to each well and incubated for 15 minutes in a dark room. The color development was stopped by adding 50 µL sulphuric acid (2 M). The absorbance (OD) was measured at 450 nm by a microplate reader (Biochrom Anthos 2020 microplate reader, UK).
3.7. Neutralization Assay
The plasmid pJFH-1 containing the full-length cDNA of JFH-1 strain (genotype 2a; Gen Bank accession no. AB047639), kindly provided by T. Wakita was used for neutralization assay. Viral RNAs were produced by in vitro transcription as previously described (18). Viruses were produced by electroporation of the viral RNAs into Huh-7 cells (19). The viral infectivity was evaluated by infecting Huh-7 cells with serial dilutions of viral supernatant. The viral titer was determined at 3 days post-infection by a focus-forming unit (FFU) staining assay. For neutralization assay, control antibody-containing anti-E1E2 MAbs AR5A (20) (kindly provided by M. Law (Scripps Research Institute, La Jolla, CA)) was used at 10 µg/mL and immune or pre-immune sera were mixed with a viral medium at 1:100 and incubated for 1 hour at 37°C. After 1 hour incubation at 37°C, the virus (150 FFU)/Ab mixtures were added onto Huh-7 cells (1 × 104 cells/well of a 96-well plate). At 6 hours post-infection, the inoculums were removed, and the cells were further incubated for 44 hours in fresh complete cell medium. Cells were then fixed and stained by immunofluorescence using anti-NS5A MAb 9E10 and neutralization effects were assessed by titration. The results were presented as the neutralization percent of virus infection compared to the pre-immune sera. The assay was performed in triplicate and the results were expressed as mean values.
3.8. Statistical Analysis
Statistical analysis and graphs were made using GraphPad Prism 7.03 for Windows (Graphpad Software Inc. 2017, La Jolla, California, USA). Data were analyzed by one-way ANOVA (Multiple-comparison HSD-Tukey test), and where necessary by Student’s t-test. P values less than 0.05 were considered statistically significant.
4. Results
4.1. Fusion Protein Modeling and Validation
I-TASSER server was used for modeling of two variants of fusion protein, NT(gp96)-E2 and E2-NT(gp96). The I-TASSER server predicted five final models for each form, and presented the best models based on such certain factors (Figure 1A). The predicted 3D structures were analyzed and validated using the ProSa-web server. The structural analysis by ProSa revealed that the Z-score value of the E2-NT(gp96) form (-3.33) was closer to the range of native proteins of similar size than the NT(gp96)-E2 form (-2.21) (Figure 1B). Therefore, according to the structural analysis results, the E2-NT(gp96) form was selected for further analysis. The secondary structure prediction by GOR4 server showed that the selected fusion protein (E2-NT(gp96)) consist of 27.4, 25.16 and 47.44 percent of alpha helix, extended strand and random coil, respectively.
 fusion protein. A, The best model for the E2-NT(gp96) fusion protein predicted by I-TASSER server. B, Z-score plot of the best model calculated by ProSaweb server. The plot shows scores of all experimentally determined protein chain currently available in the Protein Data Bank (PDB). Z-score (-3.33) of the best predicted model structure is close to the rang of scores typically found for proteins of the similar size")
Modeling of E2-NT(gp96) fusion protein. A, The best model for the E2-NT(gp96) fusion protein predicted by I-TASSER server. B, Z-score plot of the best model calculated by ProSaweb server. The plot shows scores of all experimentally determined protein chain currently available in the Protein Data Bank (PDB). Z-score (-3.33) of the best predicted model structure is close to the rang of scores typically found for proteins of the similar size
4.2. Docking Analysis
Ligand/protein complexes can exist in different conformations. The ligand-binding mode depends on the ligand conformation and the orientation relative to its receptor. One of the docking’s goals is the identification of the energetically most favorable binding pose of the ligand to the protein binding site. In order to determine the best binding mode, the docking tools generate a set of different ligand binding poses and use a scoring function to estimate the binding affinities. The Hex server determines the binding affinities with the absolute free binding energies estimation. Based on the total free energy reported by the Hex server (-818.75 kJ/mol), the E2-NT(gp96) had a high tendency to interact with the CD81 (Figure 2).
 fusion protein with HCV main receptor (CD81) using Hex server. Free energy of the fusion protein docking to the CD81(-818.75 kJ/mol) showed that the E2- NT(gp96) had high tendency to interact with CD81")
Ducking of the E2-NT(gp96) fusion protein with HCV main receptor (CD81) using Hex server. Free energy of the fusion protein docking to the CD81(-818.75 kJ/mol) showed that the E2- NT(gp96) had high tendency to interact with CD81
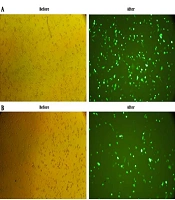
4.3. In vitro Expression Assay
To evaluate the in vitro expression of E2-NT(gp96) construct, COS-7 cells were separately transfected with pEGFP-E2-NT(gp96), pEGFP-N3 (positive control), and pcDNA3.1 (negative control) vectors. The GFP expression was fallowed at 24 hours post-transfection with a fluorescence microscope. The fluorescence microscopy revealed the proper expression of the EGFP by pEGFP-E2-NT(gp96) vector compared to the pEGFP-N3 as a positive control. In the negative control, fluorescence emission was not detected (Figure 3).
. Cos-7 cells were cultured and transfected with the pEGFP-E2-NT(gp96) plasmid, pEGFP-N3 (positive control) and pcDNA3.1(negative control). Expression of the proteins were confirmed by observation of the EGFP signal under fluorescence microscope (Nikon E200, USA) at 24 hours post transfection. A and B, GFP expression of transfected cells (before and after glinting of fluorescence) with pEGFP-N3 as positive control and pEGFP-E2-NT(gp96), respectively. No fluorescence emission could be recovered from cos-7 cells transfected with pcDNA3.1 plasmid as negative control (data not shown).")
In vitro expression assay of E2-NT(gp96). Cos-7 cells were cultured and transfected with the pEGFP-E2-NT(gp96) plasmid, pEGFP-N3 (positive control) and pcDNA3.1(negative control). Expression of the proteins were confirmed by observation of the EGFP signal under fluorescence microscope (Nikon E200, USA) at 24 hours post transfection. A and B, GFP expression of transfected cells (before and after glinting of fluorescence) with pEGFP-N3 as positive control and pEGFP-E2-NT(gp96), respectively. No fluorescence emission could be recovered from cos-7 cells transfected with pcDNA3.1 plasmid as negative control (data not shown).
4.4. Recombinant pcDNA-E2-NT(gp96) Plasmid Construction
The recombinant pcDNA-E2-NT(gp96) plasmid was generated as described in the materials and methods section. This DNA vaccine encodes a fusion protein consists of baculovirus glycoprotein 64 signal peptide (Gp64 SP), a stretch of six histidine residues (His6 tag), HCV glycoprotein E2 ectodomain, a flexible linker and N-terminal domain of gp96 (Figure 4A). The idea behind our design was that the Gp64 SP facilitates the transfer of the fusion protein to secretory pathway during translation in transduced cells and is cleaved. By this way, the His6 tag, which can be used for detection of the fusion protein in the next steps, is exposed. The integrity of the construct was confirmed by restriction enzyme digestion and sequencing. As shown in the Figure 4B, the enzymatic digestion of recombinant pcDNA-E2-NT(gp96) plasmid was shown two expected bands of size 2032 bp and 5322 bp representing the E2-NT(gp96) segment and pcDNA3.1(+) backbone, respectively. Sequencing results confirmed the accuracy of the constructed plasmid.
) cassette, and restriction enzyme digestion of the recombinant pcDNA-E2-NT(gp96) plasmid. A, Fusion gene (E2-NT(gp96)) was cloned into pcDNA3.1(+) to construct the recombinant pcDNA-E2-NT(gp96) plasmid. PCMV, cytomegalovirus immediate-early promoter/enhancer; Gp64 SP, Gp64 signal peptide; His6-Tag, a stretch of six histidine residues; E2 ectodomain, HCV E2 ectodomain; NT(gp96), N-terminal domain of gp96; BGH, Bovine growth hormone polyA signal. B, Recombinant pcDNA-E2- NT(gp96) plasmid integrity was confirmed by digestion with Nhe I/Xba I restriction enzymes. M: molecular weight, lane 1: digested pcDNA-E2-NT(gp96).")
Schematic illustration of the fusion gene (E2-NT(gp96)) cassette, and restriction enzyme digestion of the recombinant pcDNA-E2-NT(gp96) plasmid. A, Fusion gene (E2-NT(gp96)) was cloned into pcDNA3.1(+) to construct the recombinant pcDNA-E2-NT(gp96) plasmid. PCMV, cytomegalovirus immediate-early promoter/enhancer; Gp64 SP, Gp64 signal peptide; His6-Tag, a stretch of six histidine residues; E2 ectodomain, HCV E2 ectodomain; NT(gp96), N-terminal domain of gp96; BGH, Bovine growth hormone polyA signal. B, Recombinant pcDNA-E2- NT(gp96) plasmid integrity was confirmed by digestion with Nhe I/Xba I restriction enzymes. M: molecular weight, lane 1: digested pcDNA-E2-NT(gp96).
4.5. DNA Vaccine Induced Specific Antibody Responses in Immunized Mice
To evaluate the immunogenicity of the generated DNA vaccine, the pcDNA-E2-NT(gp96) plasmid was injected into the BALB/c mice. Two weeks after the last immunization, sera from the immunized mice were harvested and evaluated for specific antibodies by indirect ELISA. Three injections of the pcDNA-E2-NT(gp96) induced high titers of E2 specific total IgG antibodies in immunized mice. As expected, no anti-HCV E2 IgG was detected in sera from the control group mice, which were immunized with pcDNA3.1 (+) or PBS. As shown in Figure 5A, E2 specific total IgG level in vaccinated group (pcDNA-E2-NT(gp96)) sera was significantly higher than pre-immune sera, PBS and pcDNA3.1(+) control groups (P < 0.0001). The antibody isotypes (IgG2a and IgG1) as an indirect marker of Th1- or Th2-biased T cell responses were assessed. The recombinant pcDNA-E2-NT(gp96) plasmid injection induced high levels of both IgG1 and IgG2a isotypes with predominant IgG1. As shown in Figure 5B, IgG1 and IgG2a levels in vaccinated group (pcDNA-E2-NT(gp96)) sera were significantly higher than pre-immune sera, PBS and pcDNA3.1(+) control groups (P < 0.0001). The predominant IgG1 and IgG2a/IgG1 ratio < 1 (0.723) indicated a shift of immune responses toward Th2 for vaccinated mice.
 and isotypes (IgG1 and IgG2a) (B) levels were measured by ELISA as described in the Materials and Methods. IgG2a/IgG1 ratio calculated (the number at the top of graph) to describe the Th profile (ratios of > 1 reflects Th1 response, and the ratios of < 1 represents Th2 response). Comparison of ELISA results of immunized group with controls (pre-immune sera, PBS and pcDNA3.1(+) groups) was performed using unpaired <i>t</i>-test. Data are represented as mean ± SD. The asterisk indicates the significant difference between values determined by one-way ANOVA or Students <i>t</i>-test (P < 0.0001 denoted as ****).")
Antigen specific antibody responses induced in BALB/c mice by immunization with DNA vaccine. Specific anti-E2 total IgG (A) and isotypes (IgG1 and IgG2a) (B) levels were measured by ELISA as described in the Materials and Methods. IgG2a/IgG1 ratio calculated (the number at the top of graph) to describe the Th profile (ratios of > 1 reflects Th1 response, and the ratios of < 1 represents Th2 response). Comparison of ELISA results of immunized group with controls (pre-immune sera, PBS and pcDNA3.1(+) groups) was performed using unpaired t-test. Data are represented as mean ± SD. The asterisk indicates the significant difference between values determined by one-way ANOVA or Students t-test (P < 0.0001 denoted as ****).
4.6. DNA Vaccine Induced Strong Virus-Cross-Neutralizing Antibodies in Mice
The cross-neutralizing capacity of generated antibodies in the vaccinated mice sera with pcDNA-E2-NT(gp96) was analyzed against JFH1/HCV (genotype 2a) infectivity. To this end, sera collected two weeks after the last immunization and that of the pre-immunized mice were used. The sera from pre-immunized mice were used as a negative control. The results are presented as percent cross-neutralization of virus infection compared to the pre-immune sera. Sera of mice, which were immunized with the DNA vaccine, cross-neutralized JFH1/HCV infectivity by 55% relative to the pre-immune sera (Figure 6). As expected, sera of immunized mic with the pcDNA3.1(+) or PBS did not reduce the infectivity of the HCVcc in Huh-7 cells.

Induction of cross-neutralizing antibodies in immunized mice against HCVcc. Mice sera collected on day 0 and 56 were mixed at a 1:100 dilutions with JFH-1/HCVcc and incubated for 1 hour at 37°C, and subsequently applied to Huh-7 cells for 6 hours in triplicate. Pre-immune sera were used as a negative control. The data are shown as the percent neutralization of JFH1/HCVcc relative to the pre-immune sera.
5. Discussion
Although hepatitis C virus was discovered in 1989, efforts for the development of an effective prophylactic vaccine is ongoing. Nevertheless, the design of a vaccine that can induce a protective immunity remains a major challenge. Basically, an ideal vaccine should be capable of eliciting strong humoral and cellular immune responses (21). Among different vaccination strategies, DNA vaccines for their stability, safety, and ease of production provide a reliable approach for vaccine development (22). Although DNA vaccines can elicit both humoral and cellular immune responses, their immunogenicity is limited. In silico design and analysis of employed molecule’s properties and interactions by bioinformatics tools (9), and also simple manipulation in molecular cloning provide a desirable opportunity for improving DNA vaccines potency. In this study, we designed and generated a DNA vaccine, pcDNA-E2-NT(gp96), that encodes a fusion protein, including HCV E2 ectodomain and N-terminal domain of gp96. Two parts are fused with a flexible linker, which also provides restriction enzyme sites for molecular cloning without adding extra amino acid to the protein sequences. For this aim, we designed and fused N-terminal domain of gp96 as a biological adjuvant to N-terminal or C-terminal of HCV E2 ectodomain and produced NT(gp96)-E2 and E2-NT(gp96) fusion proteins, respectively. Then two fusion proteins were analyzed by bioinformatics tools to choose the best form for vaccine generation.
Moreover, 3D modeling of fusion proteins by I-TASSER showed that E2-NT(gp96) has better quality than NT(gp96)-E2. Local structure quality estimation showed that the residues’ charge in two forms are largely negative and stable in terms of structures. However, the estimated global accuracy of the best models of two variants indicated that E2-NT(gp96) form has correct global topology, and also it has higher confidence than NT(gp96)-E2. The evaluation of the accuracy and reliability of the predicted 3D model was done by ProSa-web. The overall quality calculated score (Z-score) of input structure is displayed in a plot that shows the scores of all experimentally determined protein chain currently available in the Protein Data Bank (PDB) (16). As shown in Figure 2A, Z-score (-3.33) of the best-predicted model structure is close to the range of scores typically found for proteins of the similar size. The evaluation of the interaction between ligand-receptor by docking is a useful method for verifying the accuracy of structural modeling. Docking is a method that predicts the conformation and orientation of the ligand at the binding site of the receptor. The association strength of the molecules is expressed by the free energy of binding (23). Free energy of the fusion protein docking to the CD81 (-818.75 kJ/mol) showed that the E2-NT(gp96) had a high tendency to interact with CD81. The highly binding affinity of E2-NT(gp96) to the CD81 receptor indicated that E2 has retained its conformation in the fusion with NT(gp96), and also fusion protein modeling was done correctly. Total results indicated that the predicted model is acceptable.
Bioinformatics analysis demonstrated that E2-NT(gp96) fusion protein had convenient features for vaccine designing. Therefore, we cloned target genes into pcDNA3.1(+) and constructed pcDNA-E2-NT(gp96) plasmid. Generated DNA vaccine potential immunogenicity was evaluated in BALB/c mice. The results of the ELISA for HCVgpE2-specific IgG measurement on sera of the immunized mice showed that the constructed vaccine induced high titer of specific antibodies. Our results are consistent with Sun and coworkers study, which they reported that immunization with pcDNA3.1 plasmids encoding E2 protein or E2-Fc fusion protein could elicit specific antibody responses in BALB/c mice (24). In another study, Deng and colleagues generated a DNA vaccine (pVRC-CE1E2) and injected into BALB/c mice. Unlike our vaccine, pVRC-CE1E2 failed to induce detectable antibody responses in immunized animals (25). The levels of isotypes (IgG1 and IgG2a) and IgG2a/IgG1 ratio showed that unexpectedly, our DNA vaccine elicited a mixed Th1 and Th2 but predominant Th2 responses. This may be due to the injection route, dose and the method of injection (26). Previous works have demonstrated that footpad administration induced stronger Th1 response than the intramuscular route we used (27, 28).
The importance of neutralizing antibodies (NAbs) in the prevention of chronic HCV infection progression is well-known (5). Thus an ideal HCV vaccine should be able to induce potent neutralizing antibody responses. As shown in Figure 6, sera of immunized mice by pcDNA-E2-NT(gp96) cross-neutralized infectivity of the HCVcc strain (genotype 2a) by 55%. Previous studies have shown that the HCV E2 glycoprotein is the main target of the neutralizing antibodies (5). Accordingly, E2 immunogenicity largely depends on its proper conformation (29). On the other hand, NAbs most often recognize tertiary or quaternary structures (30). Therefore, a vaccine platform designed to induce NAbs requires expressing the target protein in the correct three-dimensional conformation. Induction of cross-neutralizing antibodies by constructed DNA vaccine indicates that the E2 protein in the E2-NT(gp96) fusion form has presented in proper conformation to the immune system. This shows that the fusion of E2 protein to the NT(gp96) by flexible linker retains its conformation. NT(gp96) as intracellular molecular chaperone plays an important role in the folding and processing of proteins. Moreover, studies have demonstrated that gp96 or its N-terminal domain activates macrophages and dendritic cells through interaction with Toll-like receptors (TLRs) and the induction of pro-inflammatory cytokines. It also activates specific T cells by cross-presentation of peptides to MHC class I and class II molecules (31). Therefore, gp96 as a genetic adjuvant creates a potent humoral and cellular immune responses by conferring antigen presentation by both MHC-I and MHC-II.
In summary, we designed and generated a DNA vaccine (pcDNA-E2-NT(gp96)) that induced potent cross-neutralizing Abs in immunized mice. Our results showed that this recombinant plasmid might be a promising candidate vaccine against HCV infection. However, the effectiveness of the vaccine was only tested for humoral immune responses, so additional work needs to be performed for its capacity in cellular immunity induction.
