







1. Background
Von Hippel Lindau (VHL) disease is a hereditary disorder characterized by the development of benign or malignant tumors in the brain, spinal cord, eyes, adrenal medulla, kidney, pancreas, and many other organs. The disease is transmitted via an autosomal dominant pattern and has an incidence of about 1/36000 in live birth neonates (1, 2). The ophthalmologic manifestation of the syndrome, retinal hemangioblastoma, was first reported by Von Hippel, the German ophthalmologist, in 1911. In 1926, Lindau, the Swedish pathologist, described the concomitant involvement of other components of the syndrome. It was later shown that the disease resulted from abnormalities in the VHL gene located on chromosome 3 (3). The gene was cloned in 1993 (4), and further studies showed a linkage between the disease and mutations in this tumor suppressor gene. Since the first reports of the disease, more than 900 families with the disease have been diagnosed worldwide, and studies have shown that 95 to 100% of the patients have mutations in the VHL gene (5).
Advances in molecular diagnosis have led to the identification of the affected members of families at earlier stages. Improvement in surveillance of the disease and implementation of therapeutic measures has reduced morbidities and mortalities of the disease to a significant extent (6).
There are few publications focusing on the disease in the Middle East region, and little is known about the clinical and genetic characteristics of patients with VHL in the region. In this paper, we present the clinical, laboratory, and genetic characteristics of five generations of large Iranian kindred with VHL.
2. Study Population
2.1. Index Case
The proband, a 52-year-old Iranian man, was hospitalized because of headache, vertigo, and gait problems for the past two months. Medical history was positive for bilateral pheochromocytoma (BL-PCC) that had been operated on two years before admission. Familial history was also positive for BL-PCC in his son and his daughters and for retinal hemangioblastoma (R-HB) and BL-PCC in his siblings (Figure 1).

Participant pedigree
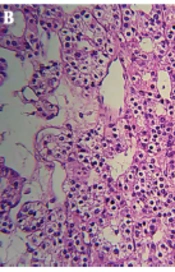
Neurological examination revealed ataxia and abnormal finger-to-nose test, while the rest of the physical examination remained unremarkable. Brain MRI showed a huge lobulated hyper-intense lesion measuring 48 by 38 by 28 mm in the right cerebellar hemisphere with significant projection into the fourth ventricle (Figure 2A). Routine laboratory tests were within normal limits, the patient was operated, and a 3 cm mass was extracted. Histopathologic diagnosis was hemangioblastoma, reticular pattern (Figure 2B). The postoperative course was uneventful, and the patient was discharged after four days.

A, Brain MRI: A huge lobulated hyper-intense lesion measuring 48 by 38 by 28 mm in the right cerebellar hemisphere with significant projection into the fourth ventricle. B, Histopathologic sample of hemangioblastoma, reticular pattern
Based on clinical findings, personal and familial history, and histopathologic characteristics of the cerebellar tumor, a diagnosis of VHL was proposed; further attention to the patient’s family history revealed that there have been multiple cases of PCC, brain tumor and retinal tumors in first and second-degree relatives of the patient. The high frequency of tumors in the patient’s family members prompted us to study his kindred to detect whether there were any other affected members. Gene sequencing of the kindred was also scheduled.
2.2. Family Members
A total of 43 members of the kindred, 24 males and 19 females, aged 2 - 77 years could be reached for evaluation. Written informed consent was obtained from all participants. In the cases of children, the consents were obtained from their guardians.
2.2.1. Data Collection
All family members underwent clinical evaluation by three endocrinologists and the geneticist of the team. Laboratory evaluations were done by using routine laboratory kits. Imaging studies, including ultrasound CT scan or MRI were carried out in affected patients. Clinical and laboratory data of patients were collected in a questionnaire designed to evaluate different clinical scenarios pertaining to the disease. Medical files and histopathology reports of patients who had been operated on before were also reviewed; affected patients were also examined by two ophthalmologists. DNA was extracted from peripheral white blood cells of all the members of the kindred. The polymerase chain reaction (PCR) was used to amplify a 292 bp fragment for Exon 3 of the VHL gene using the oligonucleotide primers (Forward: 5′-ctgagaccctagtctgccact-3′, Reverse: 5′-tctagatcaagactcatcagtacca-3′). PCR samples were analyzed by direct sequencing.
2.2.2. Diagnostic Criteria
Diagnosis of the disease was based on clinical findings (positive family history of VHL and development of a central nervous system or retinal hemangioblastoma or PCC). Diagnosis of the affected members and four cases who were unaware of their disease was confirmed by demonstration of pathogenic mutation in gene sequencing. Diagnosis of retinal hemangioblastoma was made by retinal angiography, and diagnosis of brain and spinal cord tumors was based on MRI. Diagnosis of PCC was based on elevated values of 24 hours urinary metanephrine and normetanephrine and on histopathologic evaluation of the resected tumors.
3. Result
Based on diagnostic criteria, our initial evaluations revealed that 10 members of the family had already been affected by the disease (Table 1). Among them, nine had pheochromocytoma, and one had retinal hemangioblastoma. There was no case of kidney tumors among the kindred.
| Patient No. | Age (y) | Sex | Type of Tumor | Age at Diagnosis (y) | Management | Present Status |
|---|---|---|---|---|---|---|
| III-10 | 57 | M | UL- PCC | 55 | Surgical removal, 2 years ago | Advanced CAD, UL adrenal mass |
| III-11 | 61 | M | CE-HB | 53 | Surgery, 2 times | No abnormal finding |
| III-14 | 55 | M | UL-PCC | 54 | Surgery | No abnormal finding |
| CE- HB | 37 | Surgery | ||||
| RE- HB | 50 | PH-CO | ||||
| III-16 | 52 | M | BL- PCC | 30 | Surgery | No abnormal finding |
| CE-HB | 50 | Surgery | ||||
| III-20 | 60 | M | UL-PCC, CE tumor and multiple metastases | 55 | No specific management | Slurred speech,gait disturbance bedridden, intra-cranial shunt |
| IV-5 | 28 | M | UL-PCC RE-HB | 17 | Surgery, 11 years ago | Hypertension, UL adrenal mass |
| IV-17 | 16 | M | UL-PCC | 14 | Surgery, 3 years ago | No abnormal finding |
| IV-18 | 17 | F | BL- PCC, RE-HB | 14 | Surgery, 3 years ago | No abnormal finding |
| IV-19 | 25 | F | UL-PCC, CE- HB | 23 | Surgery, 2 years ago | No abnormal finding |
| IV-23 | 35 | M | BL-PCC | 31 | Surgery, 4 years ago | No abnormal finding |
Clinical Findings of Affected Members of the Family at Our First Visit in 2017
Moreover, PCC was the most frequent tumor in the affected patients. According to patients’ medical files and our evaluations, nine out of 10 patients, aged 16 to 60 years (38 ± 17.8 years, Mean and SD) had PCC, of these, three cases had bilateral, and six cases had unilateral tumors. As shown in Table 1, cases no. IV-17 and IV-18 contracted the disease at age 14 years, and case no. IV-5 contracted PCC at age 17 years. The size of the tumors ranged from 3 to 10 cm, and histopathology was benign in all cases. There was no case of relapse or metastatic dissemination. In one patient with bilateral tumors, resection was done at one surgery, and in two cases, there was a period of 2 to 15 years lapse of time between the two surgeries. Patients number III-10 and IV-5 who had histories of PCC operation 2 and 11 years ago complained of headache and palpitation. Blood pressure was 160/105, and 170/110 mmHg, respectively, and both had palpitation and excessive sweating. Abdominal CT revealed a 7.5 by 6.4 by 4.2 cm right adrenal mass in case number III-10 and a 3.4 by 3.2 cm left adrenal mass in case number IV-10. Urinary metanephrine and normetanephrine levels were elevated in both patients. The patients were referred for surgery, and their tumors were successfully resected. Histopathologic evaluation revealed benign PCC in both patients.
Gene sequencing revealed that four asymptomatic members of the kindred (cases number, IV-7, IV-16, IV-28, and V-9) also harbored the pathogenic mutation; three of them could be reached for further evaluation and underwent ophthalmologic evaluation, MRI study of the brain, ultrasound evaluation and scanning of abdominal and pelvic cavities and measurement of 24 hours metanephrine and normetanephrine (Table 2). As shown in the table, laboratory evaluations and imaging studies of the patient no. IV-16 were negative. Evaluation of the patient No. IV-7 revealed a mass in the left adrenal gland and elevation of urine normetanephrine. The patient was referred for surgery, and 3 × 2.8 cm adrenal mass was successfully resected from his left adrenal.
| Patient No. | Age (y) | 24h Urinary Metanephrine (µg/Day) | 24h Urinary Normetanephrine (µg/Day) | Brain MRI | Abdominal CT |
|---|---|---|---|---|---|
| IV- 24 | 33 | 144 (up to 350) | 457 (up to 600) | Frontal mass and multiple spinal lesions | 1.6 by 1.7 mass in right adrenal |
| IV-16 | 16 | 160 (up to 350) | 158 (up to 600) | Normal | Normal |
| IV-7 | 13 | (59-188) | 1456 (84 - 422) | Normal | 3 by 2.8 cm mass in left adrenal |
Results of Laboratory Findings and Imaging in Asymptomatic Mutation Positive Patientsa
During the workup of patient number IV-28, an MRI of the brain and spinal cord disclosed multiple tumors. Further evaluation revealed that the patient was pregnant. An ultrasound evaluation of the abdomen showed a 1.6 by 1.7 cm right adrenal mass. Clinical examination was negative except for a pulse rate of 130/minute; her blood pressure was 110/65 mmHg, urine metanephrine and normetanephrine levels were within normal values, and imaging revealed multiple brain lesions. Treatment with prazosin was started with close monitoring for hypertension. The pregnancy was terminated at the 38th week of gestation with a caesarian section without any complications. Surgical removal of the adrenal tumor and management of CNS tumors was postponed for the post-delivery period. Considering the detection of two new cases of PCC, a total of 11 patients had PCC, among whom five cases had bilateral, while six cases had unilateral tumors.
A review of patients' medical files revealed that four patients had been afflicted with brain and cerebellar tumors in the past. The case no. III-20 had an intracranial shunt and complained of severe headache, slurred speech, and gait problems. Brain MRI revealed multiple heterogeneous enhancing masses with differing diameters (range 10 to 43 mm) in cerebellar hemispheres denoting multiple metastases (Figure 3A); reviewing the patient’s file revealed that he had a large heterogeneous mass in his right adrenal from three years ago that had not been operated because of the patient’s general condition (Figure 3B). With respect to detection of cerebellar tumor in the index case and detection of multiple CNS tumors in case no. IV-28, a total of 6 cases of the kindred had involvement of brain and spinal cord tumors.
 in cerebellar hemispheres denoting multiple metastases. B, Adrenal CT-scan, a large heterogeneous mass in the right adrenal")
A, Brain MRI: multiple heterogeneous enhancing masses with different diameters (range 10 to 43 mm) in cerebellar hemispheres denoting multiple metastases. B, Adrenal CT-scan, a large heterogeneous mass in the right adrenal
Ophthalmologic evaluation of affected patients by the ophthalmologists of the team revealed that in addition to case number III-14 who had a history of RH and photocoagulation, three more patients, numbers III-20, III-16, and IV-5 had retinal hemangioblastoma.
Gene sequencing revealed a heterozygote G to A mutation (CGG to CAG) in codon 167 of exon 3, which resulted in arginine to glutamine substitution (Figure 4); all affected subjects were positive for the same mutation. As mentioned earlier, gene sequencing revealed that four apparently asymptomatic members of the kindred also harbored the same pathogenic mutation (Table 2). As a result, our study showed that a total of 14 members of the kindred had VHL. This group was assigned for yearly clinical and laboratory surveillance. The rest of the kindred was mutation-negative and needed no further investigations.
, was found in exon 3 in the VHL gene of symptomatic and four asymptomatic family members.")
Sequence data of the amplified VHL gene; exon 3. heterozygous missense mutation (codon 39 G to A), was found in exon 3 in the VHL gene of symptomatic and four asymptomatic family members.
4. Discussion
This is the first study conducted on clinical, laboratory, and genetic characteristics of a large Iranian kindred afflicted with VHL. It shows that due to lack of knowledge on the disease and unavailability of surveillance programs, most patients were diagnosed between the fourth and fifth decades of life when the disease was markedly advanced. The study also confirms the data of other researchers on the high penetrance of the disease (about 90% at age 50 years) and shows the importance of genetic studies in the detection and management of the disease from the first decade of life.
In fact, VHL is an autosomal dominant hereditary cancer syndrome characterized by benign and malignant tumors of the central nervous system, retina, kidney, ears, adrenals, and pancreas. The disease is classified as type 1 and type 2 forms, based on the frequency of PCC. Accordingly, PCC is infrequent in type 1 patients, while PCC is almost always present in type 2 patients. Type 2 is subdivided into three forms, viz. A, B, and C. In the A form, PCC is common, but kidney tumors are infrequent. In form B, PCC is common, and kidney tumors can also be seen. Type C presents with PCC only and other components of the syndrome are missing (7). Genetic studies focusing on genotype-phenotype correlations in patients with VHL have shown that the majority of patients with type 1 disease have complete or partial deletion of the gene, while most type 2 patients have-single gene mutations (8). According to the classification provided, our patients can be classified as type 2 A.
The tendency for developing tumors in different organs seems to be secondary to inactivating mutations in the tumor suppressor gene of VHL located on chromosome 3p25-p26. Furthermore, VHL protein, along with Cullin 2, RbX1, elongin B, and Elongin C, compose the E3 ubiquitin ligase complex, which is involved in the oxygen sensing system that drives hypoxia-inducible factors alpha (HIF) destruction. VHL protein acts as the substrate recognition component in the complex that identifies hydroxylated HIF1α and HIF2α and marks it for proteosomal destruction by the E3 ligase complex. Mutations in the VHL gene with functional effects result in the establishment of HIF-1/2-α subunits, which are followed by activation of HIF target genes that are linked to tumor angiogenesis, invasion, cancer metabolic reprogramming, and metastasis (9, 10).
Mutation R167Q in exon 3 of the VHL tumor suppressor gene is the most prevalent missense mutations in patients with hereditary VHL. Estimated penetrance for the variant is over 90% by the age of 65 years, and 20% of the mutations occur sporadically. The disease can become clinically apparent from early infancy, but the mean age is around 26 years (11).
There is little information about the disease from our country. We could find only one publication on Iranian patients (12) that presented a family with malignant bilateral PCC who were diagnosed and followed as RET negative multiple endocrine neoplasm type 2 A (MEN2A) patients for more than 9 years. The family was finally confirmed as having VHL after the development of retinal tumors. Gene sequencing revealed an arginine to tryptophan substitution in codon 167 (R 167 W); two young women of the family died at the ages of 21 and 23 years from complications of severe hypertension, highlighting the importance of early diagnosis and proper management of these patients.
Pheochromocytoma followed by cerebellar and retinal hemangioblastoma was the most frequent tumor in the kindred studied. As shown in Tables 1 and 2, four patients developed PCC at the second decade of life, which shows the importance of yearly surveillance from early childhood in this syndrome. The current status of case numbers 3 - 20 (gait problem, slurred speech, and confinement to bed) underscores the importance of early diagnosis in patients with VHL.
As mentioned earlier, four cases who had the mutation were not aware of their problems until assessments revealed that two of them were afflicted; one at the age of 13 years with PCC and the other at the age of 33 years with PCC and multiple tumors of the brain and spinal cord (Table 2), emphasizing the importance of gene sequencing in the families with known cases of VHL to detect the disease at its earlier stages in the first degree relatives of affected patients.
The fact that early diagnosis reduces morbidity and mortality has led to the development of multiple surveillance programs in patients harboring pathologic mutations. Despite the minor controversy over the timing of screening in these patients, most authorities agree on beginning surveillance from the first year of life. Ophthalmologic evaluation is advised to be initiated at birth and repeated annually. Screening for PCC should begin at age two years and be continued annually. Screening for endolymphatic sac and central nervous system tumors is advised to begin at age 5 and 8 years, respectively, and be repeated biennially. Screening for renal cell carcinoma and pancreatic tumors is advised to begin at age 10 years and be continued annually (13).
4.1. Conclusions
In this study, we presented a large Iranian kindred with VHL who harbored a mutation in the VHL gene. Study results show the high penetrance of the disease and focus on the large burden imposed by the disease on the health and quality of life of patients afflicted with the disease, emphasizing the importance of surveillance from early childhood for detection and management of the disease as early as possible.
4.2. Established Facts
Von Hippel Lindau disease is a hereditary disorder characterized by the development of benign or malignant tumors in many body organs.
4.3. Novel Insights
This study showed clinical and genetic characteristics of five generations of large Iranian kindred with VHL.
