


1. Introduction
Fixed flexion deformity is a rare deformity in children. It happens due to either a bone or a soft tissue disorder. It can be associated with other joints’ deformities, multiple congenital contractures (as in arthrogryposis or Beals syndrome), or present as a part of multiple synostosis syndromes where bones are fused. Webbing of the flexor side of the elbow can be a part of either an isolated antecubital pterygium or multiple ptreygium syndromes (1). However, it has not been reported in children as an isolated deformity secondary to subcutaneous calcification in the context of pseudohypoparathyroidism. Calcinosis universalis has been rarely reported to be associated with fixed flexion deformity in childhood (2). Even in adults, diffused complicated calcinosis cutis is an extremely rare entity (3). The present case is related to a child who developed elbow joint swelling at six months of age, progressing to fixed flexion deformity and other endocrine disorders afterward, raising a suspicion of Albright hereditary osteodystrophy (AHO) that was confirmed by molecular testing. Subcutaneous calcification was limited to only one upper limb, where the fixed flexed deformity of the elbow occurred.
2. Case Presentation
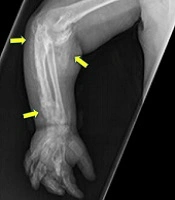
A 2.5-year-old boy from a Saudi family, who was born at term with a birth weight of 3.5 kg (32nd centile, -0.49 SDS), was referred to a general pediatrician with a history of right elbow joint swelling noticed initially at six months of age, which was followed by the movement limitation of the right upper arm, slowly progressing over the years. He had no history of trauma. At nine months of age, he was diagnosed with hypothyroidism, preceded by cold skin, dry hair, and constipation. He has been on levothyroxine since then (starting dose: 50 mcg once daily). Clinical examination revealed fixed flexion deformity of the right elbow associated with markedly limited joint movement and symmetrical hands with hyperpigmented knuckles of right metacarpal bones. Subcutaneous masses were felt along the right forearm, showing tenderness on palpation. Initial investigations showed normal corrected serum calcium (2.52, normal range: 2.20 - 2.70 mmol/L), slightly elevated phosphate (2.07, normal range: 1.33 - 1.92 mmol/L), normal ALP (308, normal range: 141 - 460 IU/L), and markedly elevated PTH (291, normal range: 21.88 - 87.55pg/mL). The X-ray of the right upper limb has been shown in Figure 1A.

A, right forearm X-ray; B, the child at the age of 9 years with the right elbow flexed deformity and hyperpigmented knuckles.
The patient was then lost to follow-up. Later, he was brought back for being examined at the age of 9 years (Figure 1B). He appeared developing appropriately for his age. Over that time between the two visits, he received oral vitamin D intermittently. His growth parameters at the second visit were height, 145 cm (98th centile, +2.07 SDS); weight, 79.1 kg (> 99th centile, +4.98 SDS); and body mass index (BMI), 37.6 kg/m2 (> 99th centile, +7.71 SDS). Biochemical investigations showed: TSH, 16.7 (0.6 - 3.8 iu/L); fT4, 11.3 (9 - 21 pmol/L); PTH, 725 (21.8 - 87.5 pg/mL); Corrected calcium, 2.45 (2.20 - 2.70 mmol/L); Phosphate, 2.09 (1.33 - 1.92 mmol/L); ALP, 308 (141 - 460 IU/L); and Vitamin D, 32 (> 50 ng/mL).
The biochemical profile indicated normal serum calcium and alkaline phosphatase levels in the context of mildly elevated serum phosphate and markedly increased parathyroid hormone. The elevated parathyroid hormone was not associated with hypercalcaemia; therefore, the diagnosis was not hyperparathyroidism. Rather, pseudohypoparathyroidism was diagnosed as there was evidence of resistance to PTH. Obviously, elevated TSH along with low-normal fT4 indicated either poor adherence to levothyroxine or its insufficient dose. Nevertheless, elevated TSH could also be due to TSH resistance. Vitamin D level was insufficient rather than deficient.
Since the child looked obese (BMI above the 99th percentile) and presented with PTH resistance, hypothyroidism, and subcutaneous calcification, genetic investigation was warranted to reach a definite diagnosis. Finally, molecular testing revealed a pathological, heterogeneous GNAS mutation (c.719-1G > A Chr20: 57484737).
3. Discussion
The main differential diagnoses of contracture in a patient presenting for the first time include chronic arthritis, post-traumatic injury, pseudoarthrosis, subcutaneous granuloma annulare, and soft tissue calcification. Chronic arthritis was assumed unlikely because of the absence of cardinal features of arthritis. Congenital forearm pseudoarthrosis is also an extremely rare condition, most likely associated with neurofibromatosis type1 (NF1) or fibrous dysplasia (4). Moreover, Cafe au lait spots in NF1 would be present in about 69% of cases with pseudoarthrosis (5). Post-traumatic injuries can lead to joint dislocation or pseudoarthrosis (6), but neither the patient’s history nor clinical and biochemical examinations were suggestive of the mentioned aetiologies for this patient’s joint deformity. Subcutaneous granuloma annulare is another rare condition, which most commonly happens in children aged two to six years old. It appears most commonly in hands, feet, ankles, and the lower parts of legs. Usually, there is a precipitating factor like an insect bite or trauma (7). It feels like a subcutaneous nodule. In this case; however, X-ray showed a radiopacity extending to the subcutaneous tissue, suggesting soft tissue calcification rather than just a granulomatous tissue as the most likely pathology. This calcification had markedly restricted joint movement. Not attending physiotherapy sessions and improper mobility of the joint leading to the development of a fixed flexion deformity. Extra-osseous calcification is categorized into three groups; a, dystrophic calcification, which is usually not associated with hypercalcaemia; b, metastatic calcification, which is resulted from hypercalcaemia; and c, heterotopic calcification related to the abnormal transformation of the connective tissue due to a complex pathogenic mechanism (8).
Obesity, hypothyroidism, parathyroid hormone resistance, and ectopic calcification, all together suggest AHO. This syndrome results from a heterozygous mutation in the GNAS gene encoding the G-stimulatory subunit (Gαs) of guanine nucleotide-binding protein, which is responsible for cyclic AMP generation, a necessary mediator for hormonal actions. Therefore, the GNAS mutations leading to the loss of either expression or function of the G-stimulatory subunit limit cyclic AMP generation, predisposing to an imprinting disorder. Maternal imprinting is responsible for the phenotypic features of the disorder as well as the multiple hormonal resistance phenotype. However, paternal imprinting results in only phenotypic features (9). Could this patient be diagnosed at an earlier age? As the reader may notice, the focus was mainly on the fixed flexion deformity caused by subcutaneous ossification, otherwise known as osteoma cutis. This itself might suggest GNAS mutations as ectopic ossification could be the first presenting feature of AHO (10). Ectopic ossification is not necessarily related to abnormal levels of calcium, phosphate, or parathyroid hormone, but it is related to the haploinsufficiency of Gαs, as it was previously described in a mouse model of AHO, where the disruption of a single GNAS allele led to ectopic ossification (11, 12).
Because subcutaneous calcification is rare during childhood, general pediatricians may not be familiar with its features and implications, leading to delayed diagnosis. However, the development of hypothyroidism, followed by pseudohypoparathyroidism, should raise the suspicion of AHO. Obesity was always there since the child was 1-year old, but it was not taken seriously as a part of the disorder; hence it was not evaluated thoroughly. Especially, the child’s phenotype was not suggestive of a specific monogenic syndromic obesity. Clinicians do not commonly evaluate obesity when it is not severe. In this case, obesity was a part of the syndrome.
After the diagnosis was made, parents were extensively counseled and informed about the importance of a regular medical follow-up. Further evaluation included CT brain and echocardiogram to rule out other possible sites of extra-osseous calcification. On further follow-up, calcium levels were noted to be low, and oral calcium supplements were administered. It was expected that the parathyroid hormone was not functioning properly due to resistance. The renal ultrasound ruled out nephrocalcinosis. Skeletal examination indicated the worsening of soft tissue calcification. Treatment with active vitamin D metabolites would be required to normalize PTH levels and to maintain normal serum calcium. However, there is still no active medical treatment to resolve established subcutaneous calcification.
Insulin resistance was evident on the oral glucose tolerance test (the hyperpigmented knuckles and creases of the back of the neck indicated acanthosis nigricans). He received a dietary regimen to reduce the dietary carbohydrate intake to tackle insulin resistance. His eating pattern remained a challenge. Maternal imprinting is usually associated with the loss of satiety, affecting the energy balance and leading to increased risk of obesity and insulin resistance. If obesity continues to become a significant problem, bariatric surgery may be considered after the completion of growth and in puberty. The patient received metformin therapy to tackle insulin resistance, and levothyroxine dose was increased to 75 µg once daily, which normalized thyroid hormones’ levels. His linear growth remained normal; therefore, the growth hormone was not started. He is still at the pre-pubertal phase, but there is a risk of delayed puberty, which may necessitate hormone therapy at an appropriate age.
There is only one previous report of a similar mutation (a de novo, novel, heterozygous, missense mutation c.719A > T in exon 10) from Hong Kong (2006), describing a girl who had developmental delay, which improved gradually using special training. Although she continued to secondary school, she had poor school performance. She was relatively obese and short with coarse facies and developed hypothyroidism at the age of 12 years, for which levothyroxine was prescribed. She remained prepubertal by the age of 13 years and showed a normal 46XX karyotype. Interestingly, the girl had no palpable subcutaneous masses or abnormal biochemical tests (13).
3.1. Conclusion
This case report described a child with subcutaneous calcification leading to the fixed flexion deformity of the elbow, identified at an incredibly early age. Obesity with insulin resistance, hypothyroidism, and pseudohypoparathyroidism raised the suspicion of AHO, which was later confirmed by genetic testing. This is the first case report of a fixed flexion deformity in a patient with a GNAS mutation in West Asia.
