


1. Introduction
Rapid-onset obesity concurrently with hypoventilation, hypothalamic, autonomic dysregulation (ROHHAD) is an uncommon disease that presents with multiorgan disorders during early childhood. The ROHHAD term and the criteria for diagnosis of this syndrome were first defined by Ize-Ludlow et al. (1) This syndrome is a distinct entity from congenital central hypoventilation syndrome (CCHS), which is presented with paired-like homeobox 2B (PHOX2B) mutation (2). ROHHAD can be seen by a variety of signs and symptoms. Since there is no single confirmatory diagnostic test, and hence late diagnosis of this syndrome leads to late intervention and eventually high morbidity and mortality, it is essential to have a high suspicion for this syndrome, especially in children over two years old with rapid-onset obesity (1, 3).
2. Case Presentation
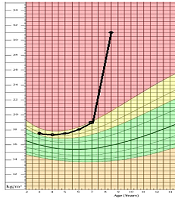
Our patient is an 8-year-old boy, first admitted to our department at age of 7 years and 8 months, with ataxia and abnormal gait from past year. The patient was born at term, from a 25-year-old mother who also has a healthy 4-year-old girl. Her mother had no complications during her pregnancy, and the infant was delivered via cesarean section for failure to progress. There is no consanguinity between his parents. He was born with a normal-range weight of 3,800 g and a birth height of 49 cm. The neonatal period was normal, and the patient had normal growth and psychomotor development. At age of five, he was admitted to another hospital because of obstruction, apnea, and snoring during sleep and went through adenectomy surgery. A year after the surgery, he was admitted again due to his somnolence during the day, but he was diagnosed only with hypothyroidism and anemia. Later, he was discharged with oral treatment of levothyroxine and folic acid, and iron. At age of 7 years and 8 months, he was admitted to our department with ataxia and abnormal gait with instability and numerous falls when he was walking. He also had shown hyperphagia, which had been resulted in 10 kg weight gain (from 31 to 41.5) in 6 months prior to his admission. With his 125 cm height, he had a BMI of 26.6. Taking a thorough medical history revealed other signs and symptoms, leading us to the final diagnosis. His BMI progression is shown in Figure 1 based on a chart from National Center for Health Statistics (4).

The patient’s body mass index on growth chart.
Six months prior to the diagnosis, he was experiencing general weakness and fatigue, and lethargy during days while doing his routine activity, forcing him to sit down and while he was playing around. He still was suffering from somnolence. It was reported that he has punched his head multiple times and has hit his head to the wall unprovokedly and without any reason. He also had gastrointestinal dysmotility and constipation from the past year. The patient had enuresis and polyuria, and urinary incontinency from the past month. In addition, the patient began to display signs of dysautonomia: a tendency to bradycardia and intestinal motility disorders (severe constipation).
His physical examinations revealed tachycardia with a heart rate of 135 bpm, normal blood pressure of 110/65 mmHg, and tachypnea with a respiratory rate of 35. He was an overweight boy with BMI of 26.6. He had central obesity with adipomastia and supraclavicular fat pad, and alongside his buffalo hump, his face was moonlike. He also was suffering from excessive sweating. His physical exam also showed central precocious puberty, with Tanner stage 4 for pubic hair and armpit hairs (Table 1). No purple striae, plethora, acne, acanthosis nigricans, or hyperpigmentation have been found. He had no alteration in pain perception nor sign of pulmonary hypertension: JVP, hepatomegaly, pedal edema. His extremities were not cold (Table 1).
| Symptoms | Age of Onset |
|---|---|
| Obesity | |
| Rapid obesity | 7 |
| Hypoventilation | |
| Obstructive sleep apnea | 5 |
| Alveolar hypoventilation | 7 |
| Autonomic dysregulation | |
| Hyperhidrosis | 6 |
| Gastrointestinal dysmotility | 6 |
| Hypothalamic-pituitary disorders | |
| Central hypothyroidism | 6 |
| Polyuria | 7 |
| Precocious puberty | 7 |
| Hyperprolactinemia | 7 |
| Neurobehavioral disorders | |
| Fatigue | 6 |
| Poor social ability | 6 |
| Aggression | 7 |
| Ataxia | 7 |
A Summary of the Patient’s Symptoms
On arrival, an arterial blood gas on room air revealed a 7.35 for pH, PaCO2 = 50.0 mmHg, PaO2 = 55.0 mmHg, and HCO3- = 24 mmol/L, and SPO2 of 83%. His laboratory investigation revealed hyperprolactinemia, while MRI of the brain and the hypothalamus and pituitary gland with contrast-enhancing revealed no abnormalities. Primary hypercortisolism was ruled out. Our complete laboratory data at the time of diagnosis are shown in Table 2.
| Parameters | Results at the Time of Diagnosis | Results at Follow-upa | Normal Range |
|---|---|---|---|
| Serum hormones | |||
| ACTH pg/mL | 13.2 | 13.1 | 7.2 - 63 |
| Prolactin ng/mL | 78 | 38.1 | 2.5 - 17 |
| Insulin-like growth factor 1 ng/mL | 322.4 | 321.7 | 72.4-323.1 |
| TSH mIU/L | 1.8 | 4.8 | 0.7-6.4 |
| T4 mcg/dL | 6.6 | 14.4 | 6.4 - 13.3 |
| T3 uptake | 29% | 29% | 24 - 39 |
| Cortisol 8 am mcg/dL | 5.9 | < 0.8 | |
| Cortisol 8 a.m. following 1 mg dexamethasone overnight mcg/dL | 0.97 | < 2.0 | |
| Vitamin D3 ng/mL | 34 | 32 | 30 - 50 |
| 24-hour Urine | |||
| Total urine volume mL/24hrs | 1700 | 1500 | 600 - 1000 |
| 24-hour creatinine mg/24hrs | 595 | 440 | 300 - 1000 |
| Vanyl mandelic acid (VMA) mg/24hrs | 5.3 | 3.2 | 54 - 138 |
| Free cortisol mcg/dL | 17.0 | 19.2 | 4.3 - 176 |
| Urine 24-hour metanephrine mcg/24-hour | 161 | 35 | 54 - 138 |
| Urine 24-hour normetanephrine mcg/24-hour | 544 | 191 | 47 - 176 |
| blood biochemical | |||
| WBC/µL | 5800 | 10400 | 4500 - 13500 |
| Neutrophil | 48% | 51% | |
| Lymphocyte | 45% | 40% | |
| Hb g/dL | 11.2 | 13.4 | 11.5 - 15.5 |
| MCV FL | 71.7 | 72.4 | 77 - 95 |
| MCH pg | 22.69 | 22.8 | 25 - 33 |
| MCHC g/dL | 31.61 | 31.5 | 31 - 37 |
| Platelet/µL | 226000 | 225000 | 140000 - 400000 |
| Blood sugar mg/dL | 78 | 103 | 70 - 115 |
| ESR | mg/dL | 14 | 10 |
| BUN mg/dL | 8.9 | 18 | 5 - 23 |
| Creatinine mg/dL | 0.95 | 0.6 | 0.7 - 1.4 |
| Bilirubin total mg/dL | 0.52 | 0.61 | < 1.0 |
| Bilirubin direct mg/dL | 0.14 | 0.15 | < 0.3 |
| Calcium mg/dL | 9.5 | 9.1 | 8.6 - 10.3 |
| Phosphorus mg/dL | 4.79 | 4.0 | 3 - 7 |
| Alkaline phosphatase U/L | 500 | 494 | 180 -2 00 |
| AST U/L | 26 | 21 | < 37 |
| ALT U/L | 33 | 27 | < 41 |
| LDH serum U/L | 417 | 571 | < 746 |
| Albumin g/dL | 4.2 | 4.2 | 3.8 - 5.2 |
| Ferritin ng/dL | 32 | 31 | 20 - 300 |
| Na mEq/L | 141 | 140 | 130 - 150 |
| K mEq/L | 3.5 | 3.5 | 3.5 - 5 |
Laboratory Findings at the Time of Diagnosis and Follow-up
Because of association of ROHHAD syndrome with Neural Tumour Syndrome, we asked for a positron emission tomography (PET-scan) alongside with chest and abdomen computed tomography scan (CT-scan). The patient was further diagnosed using Metaiodobenzylguanidine scan (MIBG scan) and a contrast-enhanced MRI scan of the brain and the hypothalamus, and pituitary gland. His 68 Ga DOTATATE PET/CT revealed several small intraperitoneal and retroperitoneal lymph nodes without significant uptake, but only metabolically inactive bilateral cervical on left axillary and intraperitoneal lymph nodes. His Abdomen CT-scan also revealed lymph nodes in celiac chain. The chest CT-scan showed that he had prominent lymph nodes. His heart echo showed mild LVH, mild diastolic dysfunction. Eye examination for Fuchs’ dystrophy of cornea and retinitis pigmentosa was normal, and he did not have strabismus.
He had repetitive apnea on a polysomnography test, so we started non-invasive ventilation for the patient. Our treatment included oral Hydrocortisone tablet, fluticasone, and salmeterol spray with salbutamol spray with montelukast tablet. For his behavioral changes, we started 10 mg fluoxetine and aripiprazole 20 mg daily. On follow-up visits, our patient did not exhibit any sodium imbalance, but he had high blood pressure, so we used captopril.
He was back to our department for configuration for his CPAP device. At this time, he was 8 years old, and he had gained another 13 kg weight and had become 54 kg with a height of 130 cm. He had a BMI of 31.95, which is +7.31 SD for his age, so we started a diet with an exercise regime. His behavioral changes improved, and he has been showing less aggressive behavior at home. He did not have any ataxia. Further, his sleep quality improved with CPAP device, and he was less somnolence during the day. However, he still suffered from autonomic dysregulation symptoms like constipation and excessive sweating. His follow-up laboratory data are shown in Table 2.
3. Discussion
ROHHAD is a complex disease with divergent signs and symptoms that needs to be in mind for diagnosis and should be treated with a high level of collaboration of various medical specialties, including endocrinologists, psychiatrists, surgeons, pneumologists, oncologists, neuropediatricians, and cardiologists, among other specialists. Early management is essential to improve prognosis and prevent serious complications, even in case of conservative treatment.
This syndrome is currently known as ROHHAD-NET because around 40% of ROHHAD syndrome also present with neural crest origin tumors e.g., ganglioneuroma, ganglioneuroblastoma; moreover, these tumors may be found years after the initial symptom (5). The underlying pathogenesis of ROHHAD is unclear, but a multitude of other genetic predisposition factors, including immunological or paraneoplastic, have been mentioned. Nevertheless, no epigenetic factors or genetic changes about this tumor have been recognized, yet there is no evidence of correlation with the autoimmune process. The theory that there may be an association between ROHHAD and paraneoplastic or autoimmune factors was first mentioned in 1995 (6). Associating with neural crest tumors highlights an immune-mediated process as in opsoclonus myoclonus ataxia syndrome (7). This theory should be considered because of the satisfactory outcomes with immunomodulatory or immunosuppressive treatments. (7, 8) This case report, in which only one of a monozygotic twins had ROHHAD, has raised doubts about genetic predispositions of this disease (9). ROHHAD patients display normal development and growth till the first symptoms arise, which happens between age of 1.5 to 9 years. Weight gain is the most common first sign, which is usually proceeded with hypothalamic dysfunction (1). The following symptoms have been reported to occur in ROHHAD patients in subsequent months and years after the initial symptoms: alveolar hypoventilation, hypothalamic dysfunction consisting of hypernatremia or hyponatremia manifested by thirst and antidiuretic hormone secretion abnormalities, diabetes insipidus, polyuria/polydipsia, hyperprolactinemia, central hypothyroidism, central precocious or delayed puberty, growth hormone (GH) deficiency, adrenocorticotropic hormone (ACTH) deficiency and in parallel autonomic dysfunction, including light-nonresponsive pupils, impaired gastrointestinal motility (constipation), body temperature disorders (hypothermia, hyperthermia), sweating disorders, reduced pain sensation, and behavioral disorders mostly irritability and aggression and fatigue and social withdrawal and poor school performances and neurological abnormalities, including seizure and blurring of consciousness (10). Although there is no confirmatory diagnostic test, the diagnosis of ROHHAD is exceptionally demanding. Hence, late diagnosis of this syndrome leads to late intervention and eventually high morbidity and mortality, it is essential to have a high suspicion for this syndrome, especially in rapid-onset obesity in children over two years old (1, 3). ROHHAD has clinical diagnostic criteria and must include rapid weight gain and hypoventilation initiated after the age of one and a half years. Patients must also have hypothalamic dysfunction with at least one of the mentioned disorders: hyperprolactinemia, rapid-onset obesity, failed growth hormone stimulation test, central hypothyroidism, electrolyte imbalances, corticotrophin deficiency, or altered onset of puberty (11). Genetic investigations should be considered to rule out other disorders with overlapping features, including CCHS and Prader-Willi syndrome. Basic cardiopulmonary, central nervous system, and neuromuscular evaluations should be performed to eliminate any chance of other diagnoses or secondary complications. Even though malfunction of the respiratory centres and their chemoreceptors is the most obvious answer for respiratory problems, scarce evidence has been published on this theory. A deficit in chemosensory function is thought to be the reason for persistent hypoventilation; however, Carroll et al. (12). explained that the responses of ROHHAD patients to hypoxia and hypercarbia are similar to those of healthy young adults. Yet, decreased inspiratory drive and tidal volumes during some stimuli concurring with a lacking behavioral perception of asphyxia make these patients prone to hypoxemia and hypercarbia (12).
3.1. Conclusions
Our case indicates that ROHHAD is a complex disease with divergent signs and symptoms that needs to be in mind for diagnosis and should be treated with high level of collaboration of various medical specialties. Since late diagnosis of this syndrome leads to a high chance of morbidity and mortality, it is vital to pay extreme attention to this syndrome. This is even more critical for children over 2 years old with rapid-onset obesity, which is accompanied by other symptoms. Here, the patient’s complaint was ataxia which, revealed the underlying cause after investigation.
