



1. Introduction
Paragangliomas (PGL) are rare extra-adrenal neuroendocrine tumors that can form within sympathetic and parasympathetic ganglia throughout the body (1). Although PGLs arise from neural crest-derived chromaffin cells, individual tumors differ with regards to location and malignant potential. PGL are genetically heterogeneous tumors that have the highest degree of heritability of any endocrine tumor type (2). Germline mutations in a variety of genes, including succinate dehydrogenase subunit B (SDHB), von Hippel Lindau (VHL), neurofibromatosis type 1 (NF1), and RET have been associated to give rise to the tumors (3).
RET is a 21-exon proto-oncogene located on chromosome 10 that has been associated with the multiple endocrine neoplasia type 2 (MEN-2) syndrome. Individuals with RET mutations leading to MEN-2 are at risk for developing hyperparathyroidism, medullary thyroid cancer, and pheochromocytomas/paragangliomas (4). The University of Utah MEN-2 and RET genetic database has documented approximately 200 different mutations, most of which have not been well characterized (5). Here, we describe a patient with a history of parathyroid adenoma who developed a pancreatic retroperitoneal paraganglioma and was found to have a RET mutation variant of unknown significance. This particular variant has not been previously documented in the University of Utah MEN-2 and RET genetic database, making its clinical significance unknown.
2. Case Presentation
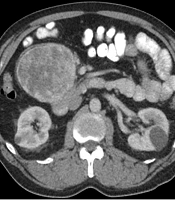
A 64-year-old Caucasian male originally presented to his primary care physician due to gradually increasing right hip pain for more than two months. He denied any significant weight loss, night sweats, heart palpitations, or other systemic symptoms. His past medical history was significant for a parathyroidectomy with resection of a single parathyroid adenoma and well-controlled arterial hypertension. Family history included prostate cancer in his father. On exam, the patient had reproducible right hip pain with passive and active motion. Due to concerns for musculoskeletal pathology, magnetic resonance imaging (MRI) of the spine was obtained and had demonstrated mild degenerative changes in the spine and an 11.2 cm mid-abdominal retroperitoneal mass. Contrasted enhanced computed tomography (CT) of the abdomen and pelvis was obtained to characterize the mass; this showed an 11 cm ovoid, solid, enhancing mass in the right mid-abdominal mesentery with no associated lymphadenopathy (Figure 1). Given a low suspicion for a catecholamine-producing tumor (which would have necessitated further workup), the decision was made to proceed directly to an image-guided biopsy to establish a diagnosis and guide treatment. The biopsy demonstrated an atypical epithelioid and spindle cell neoplasm. The patient underwent an elective exploratory laparotomy for removal of the mass. Intraoperatively, the tumor was found to be adherent to the head of the pancreas and to the confluence of the superior mesenteric and portal veins. The tumor was resected en-bloc with a margin of pancreatic tissue and the portal vein defect was repaired primarily.

CT of the abdomen showing a large peri-pancreatic mass abutting the mesenteric vessels.
Histologic evaluation of the tumor showed evidence of neoplastic cells arranged in nests and trabeculae within the tumor’s vasculature. Round to oval cells with moderate to abundant eosinophilic granular cytoplasm were found within the tumor bulk. Tumor was metastatic to one of the lymph nodes. The tumor immunohistochemistry demonstrated a Ki-67 index < 1%, positive succinate dehydrogenase subunit A (SDHA), SDHB, chromogranin A, and S-100 stains, (Figure 2) and negative cytokeratin CAM 5.2 and human melanoma black-45 (HMB45) stains. These findings were consistent with a PGL with metastasis to the lymph node.

Immunohistochemical staining showing positivity for synaptophysin, chromogranin A, and S-100 consistent with paraganglioma. Ki-67 is low at < 1%.
Once the diagnosis was obtained, urine catecholamines were immediately measured. The patient had elevated urine metanephrines 382 mcg/24 h (normal range 44 - 261 mcg/24 h) and urine normetanephrines 650 mcg/24 h (normal 138 - 521 mcg/24 h) demonstrating a functional PGL. Gallium-68 DOTATE scan showed no evidence of distant metastatic disease. Genetic analysis was performed on a peripheral blood sample using a hybridization-based protocol to enrich for regions of interest. Amplicons from the coding exons of 34 genes of interest were created using real time polymerase chain reaction, and subsequently sequenced using Illumina® technology. When compared to a reference sequence, a RET gene variant of unknown significance [c.731C>T (p.T244I)] was identified. Screening thyroid ultrasound did not demonstrate any suspicious thyroid nodules. At his three month follow up, our patient reported full recovery from the surgery with no further symptomatology and his metanephrines had normalized. At his one-year follow-up, he reported he was doing well. However, a suspicious left renal mass, concerning for renal cell carcinoma, was seen on his screening CT abdomen and pelvis. There was no evidence of recurrent disease from his metastatic PGL on imaging. At this time, patient is undergoing further work-up with urology.
3. Discussion
Germline mutations of the RET proto-oncogene are associated with MEN-2 syndrome and familial medullary thyroid carcinoma (MTC). The penetrance of MEN-2 varies according to the specific causative mutation (6). RET gene mutations have also been associated with lung cancer, adenocarcinoma of the colon, and melanoma (7). In patients with diseases associated with the RET gene mutation, amplicons of genomic DNA can be sequenced and compared to reference data to identify specific mutations. Exons 5, 8, 10, 11, 13, 14, 15, and 16 are specifically screened for the presence of RET mutations (8).
Our case describes a patient with no familial history of genetic syndromes and previous parathyroid adenoma who presented with a pancreatic paraganglioma associated with a RET T244I germline mutation. Since mutations in the RET gene can result in a wide range of presentations, attempts have recently been made to catalog the phenotype of individual mutations for prognostic purposes. This mutation, as of yet, is not listed under the University of Utah MEN-2 and RET genetic database. The mutation has, however, been reported once in ClinVAR database as a RET variant of uncertain significance (9).
Mutations in exons 10, 11, 13, 14, 15, and 16 of the RET gene are clinically relevant as they are associated with MEN syndromes (10, 11). The development of MEN-2A has largely been linked to mutations in exons 10 and 11, coding for the cystine-rich extracellular domain of the tyrosine kinase (codon 634 in exon 11 is the most commonly altered) (12). Conversely, in MEN-2B, a point mutation in exon 16 (Met918Thyr, M918T) that results in a conformational change in the intracellular binding pocket is responsible for 95% of hereditary cases (10). In our patient, the point mutation was found in exon 4, which codes for a portion of the extracellular domain of the RET gene. A review of the University of Utah MEN-2 database has not demonstrated any mutations responsible for MEN syndrome occurring in this codon (5). Recently, Zhang et al identified two separate mutations in exon 4 that were thought to be linked to familial medullary thyroid carcinoma in an eastern Chinese population (13).
It is unclear if our patient has MEN-2A syndrome. Given his history of parathyroid adenoma and paraganglioma, our patient may have a pathogenic variant. MEN syndromes are mainly inherited in an autosomal dominant fashion, however the probability of de novo pathogenic variants to cause MEN-2A is approximately 5% (14). The new finding of the renal mass concerning for renal cell carcinoma may suggest that the patient have some variant of MEN syndrome. RET gene is altered in 1.25% in renal cell carcinoma patients (7). Further urological work-up may assist in determining the genetic syndrome our patient harbor. Given our patient’s unique presentation, his case highlights the importance of documenting phenotypic presentations of newly described mutations as it not only helps in determining prognosis factors, but also guide in clinical surveillance.
The clinical course of a patient with PGL is variable, with survival most notably depending on the malignant potential of tumor. Like other neoplasms, PGLs can be considered malignant if distant metastases are present. One study found that, on average, malignant PGLs reoccurred at a distant site roughly 5.5 years after the time of initial diagnosis. Such patients with distant metastases had a median overall survival of 24.6 years (15). Determining the malignant potential of a solitary tumor, however, remains difficult. The Grading System for Adrenal Pheochromocytoma and Paraganglioma (GAPP) was developed to predict potential aggressive behavior of PGL (16). This grading system showed that a histologic pattern of large, irregular sized nest or pseudo-rosette forming cells, high cellularity, the presence of coagulation necrosis and vascular invasion, Ki-67 immunoreactivity > 3%, and norepinephrine producing PGL were associated with malignant potential. Patients with malignant PGL were also found to have rapid disease progression if associated with male sex, older age at diagnosis, synchronous metastasis, large tumor size, elevated dopamine, and not undergoing resection of the primary tumor (15).
Using the GAPP score, our patient would have a low to intermediate risk with a metastatic rate of 3.6 to 60% and five-year survival of 66.8 to 100%. Considering the likelihood the patient has renal cell carcinoma, we would expect his five-year survival to be less than expected according to the GAPP score.
Given the morbidities associated with PGL’s and other RET-derived tumors, it is recommended that an affected patient’s family members also obtain genetic testing to determine if they have any somatic RET mutations. Patients and family members with RET mutations will benefit from annual biochemical screening, including serum calcium, carcinoembryonic antigen, calcitonin, 24-hour urine metanephrines, and vanillyl mandelic acid (VMA) levels. Patients should also undergo screening ultrasound for thyroid cancer.6
We recommend that patients be followed every four months in the first year after surgery. Afterwards, patients should be seen semi-annually to annually for the following two years, and then annually thereafter. Our recommendation is similar to that of the National Comprehensive Cancer Network (NCCN), which recommends patients have semi-annual follow up for three years after surgery, and then annual follow up for up to ten years. After ten years, NCCN recommends that physicians consider surveillance as clinically indicated (17). We recommend that patients (regardless of mutational status) should undergo life-long surveillance since recurrence and metastatic disease can present years after from the primary diagnosis (15).
