
Fulltext
Rickets is usually a result of vitamin D deficiency, secondary to lack of oral intake of calciferols or inadequate exposure to the sun. In rare circumstances, the disease develops despite adequate vitamin D supplementation, a condition named “vitamin D resistant” rickets. Hereditary vitamin D resistant rickets (HVDRR), formerly referred to as vitamin D dependent rickets type 2, is an uncommon form of bone mineral abnormality, usually encountered in early childhood. In most cases the disease is associated with alopecia. Pattern of inheritance is autosomal recessive and males and females are affected with equal frequency. In the majority of such patients, mutation in vitamin D receptor (VDR) gene leads to formation of mutant receptors (MRs) with decreased functional abilities (1-3).
VDR is a member of the class 2 nuclear hormone receptor superfamily that includes thyroid, steroid, and retinoid acid receptors. Similar to other members of the family, VDR has a carboxy-terminal named ligand binding domain (LBD) that binds with 1-25(OH) 2 D3 and a DNA binding domain (DBD) that interacts with specific segments of DNA. To exert its physiological effects, the receptor needs to form a heterodimer complex with the retinoid x receptor (RXR) and subsequently bind with specific sequences in DNA molecule, named vitamin D response elements (DRE). Binding to these sites and completion of the effects depends on the interaction of substances named transcriptional coactivators. Bone mineralization abnormality, the hallmark of the disease, results mainly from defective intestinal absorption of calcium, secondary to unresponsiveness of the mutant receptors of the gastrointestinal tract to 1-25 (OH) 2 D3 (4, 5).
HVDRR was reported for the first time by Brooks et al. in 1978 (6). In 1988, the human VDR gene was cloned by Baker et al. (7) and the first VDR gene mutations in 2 patients with HVDRR was reported by Hughes et al. (8). The disease is rarely encountered and the literature review showed that the reported cases in medical literature barely surpass 100. Detection of different mutations by gene sequencing and studies on the functional characteristics of different mutant receptors (MRs) shed more light on the etiology and pathogenesis of the disease.
The current paper presented the clinical and laboratory characteristics of 2 Iranian siblings with HVDRR and alopecia, followed up for over 27 years.
All subjects gave their informed consent before participating in the study, and the study observed the tenets of the declaration of Helsinki.
The patients, a brother and a sister born to a consanguineous marriage, were first visited by one of the authors (AAG) in 1988. The male sibling was 6 years old and the female one 3. The parents were first-degree cousins. Main problems were growth retardation and difficulty in walking. The patients were diagnosed with nutritional rickets and the male and female siblings both had 16 and 25 injections of vitamin D3 respectively (each containing 600,000 units of cholecalciferol) with apparently no improvement. On the examination, both had typical features of rickets, including widening of wrists, prominent rickets rosary, and difficulty in walking. The male sibling was 101.5 cm tall and the female 80 cm, both under the third percentile. Bone age was retarded in both. The clinical presentation of rickets was more prominent in the female sibling, who also had a prominent genu varum. Both patients had alopecia totalis. Eyebrows and eyelashes were absent in the male sibling. Values of serum calcium, phosphorus, alkaline phosphatase (ALP), and parathyroid hormones (PTH) are shown in Table 1. Results of the routine laboratory evaluation including serum albumin were normal. Evaluation of serum 25 OHD and 1-25 (OH) 2 D3 was not available at that time.
| Male | Female | Reference Range | |
|---|---|---|---|
| Calcium | 8.8 | 7.3 | 8.6 - 10.3 mg/dL |
| Phosphorus | 4.2 | 3.9 | Children = 3.5-6 mg/dL, adults = 3.5-5 mg/dL |
| Alkaline phosphatase | 635 | 2073 | Children = 230 - 480 IU/L, adults = 120 - 230 IU/L |
| PTH | 72 | 70 | 15 - 60 pg/mL |
A clinical diagnosis of HVDRR was proposed. Treatment was started with calcitriol 1 µg/day and the dose was gradually increased to 8 µg/day. Oral calcium was also advised and increased to 1 to 3 g/day. Despite high doses of calcium and active vitamin D, no significant improvement was observed in the patients’ condition. With normalization of serum calcium and ALP in the male sibling at around the age of 12 years, the medicine was discontinued. Treatment was continued for the female because of abnormal laboratory values and severe genu varum. At the age 14 years a corrective bilateral dome shape femoral and upper and lower tibial osteotomy was done for the female sibling. Both lower limbs were fixed by pin and plasters in well alignment position for 12 weeks. After surgery, lower limbs were supported by long leg brace for 3 years. At the age of 16, with normalization of laboratory values, the drugs were discontinued for the female sibling as well. The patients were visited yearly and their laboratory values were evaluated (Figure 1), revealing normal values despite cessation of the drugs. Final height of the male sibling was 165 cm. He is recently married and has a normal appearing son. Final height of the female sibling is 146 cm. She completed puberty and has regular menstrual cycles.

In their last visit, November 2015, the patients were reevaluated. Serum levels of 1-25(OH) 2 D3 were > 210 pg/mL in both (normal range, 23 to 76 pg/mL), values were 93 pg/mL for the father and 117 pg/mL for the mother respectively. Bone mineral density (BMD) assessment in female sibling showed a Z score of 0.8 in femoral neck and -0.1 in lumbar spine, and the male siblings Z score was -2.9 for the femoral neck and -4.1 for the lumbar spine; Z score for the 1/3 distal radius was -3 and -9.8 in the female and male siblings, respectively.
Peripheral blood samples were collected for genetic analysis from the patients and their parents after written informed consent. Genomic DNA was isolated as previously described (9). The coding and flanking regions of the VDR (exon 4 to 11) were amplified by polymerase chain reaction (PCR) and the products were analyzed by direct sequencing. Sequencing analysis revealed a homozygous G to A mutation in the DNA binding domain (DBD) of exon 4 (g.30994G > A) in both siblings (Figure 2). The mutation had changed the amino acid number 26 from valine to methionine. The parents were also heterozygote for the same mutation.
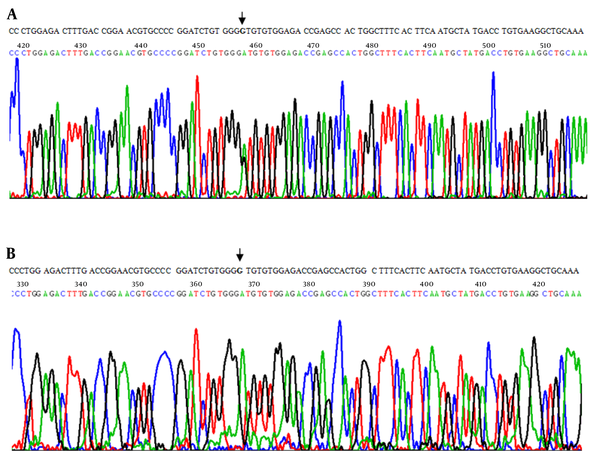
The forward sequence revealed the GTG > ATG (Val26Met) mutation in the heterozygous state in both parents (A) and in the homozygous state for the mutation in the patients (siblings) (B). The top row represents the normal sequence and arrows indicate the point mutations.
The current paper presented clinical, laboratory, and genetic characteristics of this unusual form of rickets in 2 Iranian siblings who were under observation for more than 27 years. The patients are the second and third cases of HVDRR that were genetically studied in Iran (10).
Clinical presentation of the patients was classic; both had growth retardation and difficulty in walking since early childhood, but the severity of the disease was more pronounced in the female case. Growth chart of the male sibling showed that growth velocity improved with onset of puberty and increased from below the 3rd percentile to the 10th percentile, while the female sibling remained under the 3rd percentile line during and after completion of puberty. It seems that the severe growth retardation in the female sibling might have been related in part to greater aberrations in functional abilities of her receptors.
Severity of alopecia in the patients remained unchanged during the follow-up period (Figure 3). Pathogenesis of hair loss in HVDRR is not yet fully elucidated. It seems that normal hair growth depends on interaction of VDR with a 130-kDa nuclear transcription factor named hairless protein (Hr). Apparently, formation of the heterodimer from the 2 is crucial for completion of the hair growth cycle. In patients with HVDRR, abnormalities in VDR structure prevent the formation of the designated heterodimer and hinder completing the cycle (11). Alopecia in HVDRR patients does not respond to any treatment protocols.

Data of bone mineral density was significantly different in the siblings. As stated earlier, while the male sibling had severe bone loss, the female sibling had normal BMD, an unexpected finding because the female sibling had a more severe form of the disease based on clinical (Genu varum and the need for corrective surgery) and the laboratory findings (low calcium and sky high ALP); the discrepancy can be attributed to the fact that factors other than vitamin D receptors are implicated in bone accretion. Data of the male sibling were very different from those of Tiosano et al. and Damiani et al. who reported that BMDs of their patients at post-pubertal age were not different from that of the normal population (12, 13).
Laboratory data of the siblings differed significantly; in the male sibling, serum calcium was at lower limits of normal or mildly lower and serum phosphorus was normal, while the values for the female sibling revealed very low serum calcium and elevated values for ALP. Contrary to majority of published cases reporting low serum phosphorus, these values were normal in the male sibling and minimally decreased only in one occasion in the female. In fact, the serum phosphorus values were higher than normal in the female sibling during childhood and adolescence. Values for serum PTH only minimally elevated initially in both cases and the female sibling had normal PTH values despite low serum calcium during the follow-up period. authors had no explanations for serum phosphorus and PTH levels in the patients. As expected, the serum level of 1-25(OH) 2D3 markedly elevated in both siblings. The higher than normal values of 1-25(OH) 2D3 in the parents was a physiologic compensatory response to their heterozygous state.
Treatment of rickets and hypocalcemia is challenging in most cases. Despite the fact that mutant receptors were unresponsive to 1-25 (OH)2 D3, various forms of vitamin D (cholecalciferol, ergocalciferol or calcitriol) were used in these patients with doses up to about 100 times above normal (14, 15). Calcium was also used with different doses in all cases. Response to treatment varied with the improvement in a limited number of cases (16). In the majority of cases, including the current study ones, no significant improvement was gained despite pharmacologic doses of vitamin D and oral calcium. Long-term intravenous calcium infusion, via a central line, was the most effective treatment used with success in some cases (17). Improvement of rickets in these patients, after normalization of serum calcium, points to the fact that vitamin D exerts its effect on bone synthesis solely by increasing intestinal calcium absorption and its direct effect on bone is minimal, if any.
Spontaneous remission of the disease with normalization of serum biochemistry was reported in most cases. In the majority of cases it happened around pubertal age and patients had normal laboratory values for years despite cessation of calcium and vitamin D treatment. With respect to the fact that defects in mutant receptors and their functional abilities are permanent, the improvement in serum chemistry points to non-VDR dependent pathways for intestinal calcium absorption. Animal studies showed that VDR knockout mice could maintain normal levels of calcium and PTH during pregnancy and lactation by increasing intestinal calcium absorption through vitamin D independent mechanisms. The effect was attributed to increased levels of human placental lactogen and estradiol during pregnancy. The role of estrogen was later endorsed by the fact that the hormone upregulates expression of intestinal calcium absorption genes (18). Further studies showed that parietal cells of the intestinal wall have potent aromatase activity and the ability to synthesize estrogen locally (19). In a study on the intestinal calcium absorption in the patients with HVDRR of different age groups, Tiosano et al. showed that intestinal calcium absorption was highly vitamin D dependent from early childhood until completion of puberty. They also showed that fractional calcium absorption (FCA), which is low in early years of life, increases significantly after completion of puberty. It was also shown that bone calcium absorption increases to values significantly greater than normal controls in patients aged 18 to 26 years (12). As a result, it seems that high levels of sex steroids in the post pubertal period lead to expression of genes responsible for elaboration of proteins involved in intestinal calcium absorption via vitamin D independent pathways (19).
Based on the studies on the functional abilities of mutant receptors, it was found that VDR gene mutation was the underlying mechanism of the disease in the majority of reported cases. Missense mutation was the most common type but other forms such as nonsense mutation, frameshift and splice site mutations were also reported in a few cases (4). The disease was also associated with compound heterozygote mutation or mutation in the RXR gene (4). Mutations were found both in LBD and DBD. In patients with mutations in LBD, binding to 1-25 (OH) 2 D3 and further steps in activation of the hormone-receptor complex, such as heterodimerization with RXR and recruitment of the coactivators are abolished. In patients with mutations in DBD, hormone-binding ability is retained, but the activated receptor cannot bind to designated DRE and proceed to completion of the expected effects.
The G to A mutation in VDR gene, which changes amino-acid number 26 from valine to methionine, was reported in only 1 case (3). The patient was a 13-month-old male, result of a consanguineous marriage who presented with macrocephaly, convulsion, clinical symptoms of rickets, partial hair loss, and failure to thrive. His laboratory evaluations revealed low serum calcium and phosphorus with high serum alkaline phosphatase, PTH, and 1-25 (OH) 2 D3. Due to unresponsiveness to oral calcium and high doses of active vitamin D, the patient was treated with IV calcium for about 8 months, which led to improvement in his blood chemistry and healing of bone abnormalities.
The amino acid valine at position 26 is a highly conserved amino acid and is located in the first zinc finger of the DBD region of VDR. Studies of functioning abilities of the mutant receptor showed that V26M mutation binds normally to 1-25 (OH) 2 D3. The mutation also does not affect RXR heterodimerization or interactions with coactivators or corepressors. Resistance to 1-25 (OH) 2 D3 is the result of failure of the binding of the receptor to the vitamin D binding elements of DNA and activation of the responsible genes.
In conclusion, the current paper presented the clinical and laboratory characteristics of 2 Iranian siblings with a rare disorder. It also presented the changes in clinical and laboratory features of the disease over 27 years of surveillance.
