


1. Introduction
Although the adrenal glands contain no lymphoid tissue, secondary adrenal lymphoma is responsible for about 4% to 5% of all non-Hodgkin lymphoma (NHL) cases (1). On the other hand, primary adrenal lymphoma (PAL) is rare and accounts for less than 1% of all extranodal lymphomas (2). The majority of patients with PAL have bilateral disease at diagnosis unlike secondary involvement which is usually unilateral (3). The most prevalent subtype of PAL is diffuse large B-cell lymphoma, which affects roughly 70% of patients (3, 4). Primary adrenal lymphoma typically affects elderly patients, involving bilateral adrenal glands with an average diameter of 8 cm. In addition, the presence of an extra-adrenal disease at the time of diagnosis is uncommon (5-7). Because of its generic signs, the diagnosis of PAL is usually difficult and pathological examination is the only approach to establish the diagnosis (8). The role of surgery for the management of PAL remains controversial and has been associated with poor prognosis (3). Generally, PAL has poor prognosis, with a 17.5 percent one-year survival rate (3).
Here, of 17 patients diagnosed with adrenal lymphoma in our hospital, we represent the most two challenging cases where the diagnosis was confirmed only after adrenalectomy; one primary and one secondary, in addition to including a mini-review of the literature.
2. Case Presentation
2.1. Case Report 1
Sixty-seven years old male patient presented with a 5-week history of nausea, weight loss, and epigastric ache. The patient was HIV positive and had a past medical history of ulcerative colitis, asbestos exposure and moderate smoking. Computerized tomography (CT) abdomen showed bilateral enlarged adrenal glands up to 6 cm, they appeared well defined and homogeneous (Figure 1). On magnetic resonance imaging (MRI) scan, on T1 the adrenal glands appeared relatively homogeneous and of intermediate to low signal with no high signal acute haemorrhage. While, on T2-weighted and STIR, they showed no low signal macroscopic fat on STIR and no high signal cystic areas. WBC level was 3.8 × 109/L, Hb level was 9.1 g/dL and the PLT count was 268 × 109/L. Kidney and liver function tests were unremarkable. FSH, LH, Prolactin, Testosterone, serum SHBG, plasma metadrenaline and normetadrenaline levels all were within the normal limits. Histology from core biopsies was inconclusive. After discussion in the endocrine MDT, the decision was for adrenalectomy. The patient underwent laparoscopic right adrenalectomy and the final histology reported sections showing adrenal gland infiltrated by diffuse large B-cell lymphoma. The tumour cells were positive for CD20 and negative for CD3, Bcl6, CD5, CD10, AE1/AE3 and Bcl2. The proliferation rate, as assessed by Ki67, was in a range from 90 - 95%. Post-operatively, the patient was referred to the haematology team for chemotherapy with CHOP regimen. Rituximab was omitted as there was a concern that the patient was already significantly immunosuppressed from the treatment for his ulcerative colitis. Follow-up CT scan showed enlarged aorto-caval and coeliac axis lymph nodes with probable new lymphoma deposits in the right kidney and liver. Three months later, the patient had a large solitary supra tentorial left frontal parenchymal mass lesion measuring about 4 cm, which was confirmed to be metastatic diffuse large B-cell lymphoma. The patient passed away after two months.

Computerized tomography of abdomen showing significantly enlarged both adrenal glands. They are well defined and homogeneous. Maximum diameter is about 6 cm.
2.2. Case Report 2
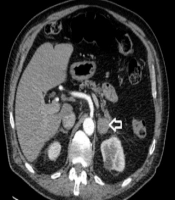
Seventy-seven years old male patient diagnosed 3 years earlier with a diffuse large B cell non-Hodgkin’s lymphoma and was treated with chemotherapy. Past medical history included type II diabetes and DVT. The patient presented with abdominal pain and altered bowel habit. WBC level was 6.1 × 109/L, Hb level was 121 g/L, PLT count was 173 × 109/L, kidney and liver functions tests were unremarkable and the adrenal hormonal profile was within the normal limits. CT abdomen was performed and showed slightly irregular enhancing mass measuring 26 × 21 mm in the left adrenal gland (Figure 2). PET-CT scan revealed increased FDG uptake in the left adrenal gland. Computerized tomography guided biopsy was arranged and showed that the left adrenal gland tumour further increased in size to 52 × 41 mm. The mass was showing venous invasion extending into the left renal vein confirming a malignant tumour and there was also involvement of the capsule of the anterior aspect of the left kidney (Figure 3). The tissue biopsy was inconclusive and showed atypical lymphoid infiltrate composed of medium to large sized atypical lymphoid cells showing occasional mitotic figures and numerous apoptotic bodies. Immunohistochemistry staining was positive for CD20 (small and large cells) and CD3 T-cells. The case was discussed in the endocrine MDT. The patient underwent radical nephrectomy and adrenalectomy with enbloc distal pancreatectomy and splenectomy. The final histology showed relapsed diffuse large B-cell lymphoma in the form of diffuse neoplastic lymphoid infiltrate composed of polymorphic centroblastic cells admixed with abundant small lymphocytes involving the adrenal gland, renal artery and perirenal fatty tissue associated with extensive necrosis. Immunohistochemistry staining was positive for CD20, CD3+ T-lymphocytes, MUM1 and bcl2, and negative for CD10, bcl6, and CD5. The Ki67 index was about 80%. Although the patient was commenced on R-CHOP regimen, he had a complicated course of recovery with intracranial involvement by the disease and passed away within three months of surgery.

Computerized tomography of abdomen showing a 27-mm left adrenal nodule of a 77-year-old patient with previous diagnosis of diffuse large B cell non-Hodgkin’s lymphoma

Follow-up computerized tomography of abdomen showing the left adrenal gland tumor measuring 52 × 41 mm and invading the left renal vein and the capsule of the anterior aspect of the left kidney
3. Discussion
Lymphoma arising in, and confined to, the adrenal glands is termed PAL. Primary adrenal lymphoma is a rare condition with only about 100 cases reported in the previous medical literature (9). On the other hand, lymphoma involving the adrenal gland on a background of disseminated lymphoma is considered a secondary lymphoma. In an autopsy research, Rosenberg et al. found that the adrenal glands were involved in 24% of those with disseminated NHL (1). Most patients with PAL have bilateral adrenal glands involvement, unlike those with systemic lymphoma, which is usually unilateral (5). Males are more affected than females, having a 7: 1 male to female ratio, and the mean age of diagnosis is 70 (10). Seventeen patients were diagnosed with adrenal lymphoma in our hospital. The average age of diagnosis was 70.7 ± 13 years of age, while male to female ratio was 9: 8. Two cases only required adrenal surgery. Of the two cases, the first patient was diagnosed with a primary adrenal lymphoma, while the second one had secondary lymphoma as he was diagnosed with B-cell lymphoma 3 years earlier.
There is no lymphoid tissue in the human adrenal glands, and the exact pathogenesis of primary adrenal lymphoma remains unclear. Many factors have been involved including immune dysfunction, mutations in the p53 and c kit genes, autoimmune associated infections like Epstein Barr virus and HIV infections (5, 10, 11).
The diagnosis of primary adrenal lymphoma is usually challenging because most of the symptoms are non-specific. The disease is characterized by bulky involvement of the gland with a median maximum dimension of 8 cm (4 - 17 cm). Local symptoms like lumbar pain may be present, as well as systemic signs such vague abdomen pain, anaemia, weight loss, unexplained fever, hypercalcemia, and thrombocytopenia (4, 6). Only 50% of individuals have concurrent adrenal insufficiency, which occurs only after the adrenal glands have been destroyed by at least 90% (12). Vomiting, extreme fatigue, skin discoloration, and hypotension are all signs of adrenal insufficiency (13).
Imaging techniques being used for the diagnosis of adrenal lymphoma include ultrasonography (US), CT, MRI and functional imaging, like gallium 67 scintigraphy imaging or positron emission tomography scans (14, 15). On CT scans, primary adrenal lymphomas are characteristically bilateral, variable in density, often with areas of necrosis and/or haemorrhage. Lesions with homogeneous density also have been reported (16). On T1-weighted magnetic resonance (MR) images, these lesions show low-signal intensity, while on T2-weighted images they show high-signal intensity, with occasional areas of mixed signal (17). Despite this, there is no pathognomonic appearance on CT or MRI to identify PAL, and distinguishing PAL from metastatic lesions is challenging.
Only histological evaluation of tissues acquired by CT, ultrasound guided biopsy or surgical excisional biopsy utilising a laparoscopic approach can confirm the diagnosis of primary adrenal lymphoma. Fine-needle aspiration is generally not recommended due to the high rate of false results. More than 80% of PAL is diffuse large B-cell lymphoma (5). Uncommon variants include anaplastic large cell, angiotropic/intravascular and T cell (18-20). The entire 17 patient in the current study were diagnosed with diffuse large B-cell lymphoma.
CNS involvement has been documented in 5% of aggressive intermediate-grade NHL patients, either initially or at relapse (21). A high serum LDH level (22), an intermediate to high-risk International Prognostic Index (IPI) (23), and involvement of more than one extranodal location, including the marrow (24), are all risk factors. Our two represented patients had CNS involvement by lymphoma which contributed to their outcomes.
There are different treatment modalities for the management of PAL which include chemotherapy, bilateral adrenalectomy, radiotherapy, and a combination of these (12). The role of surgery is still controversial and has been associated with poor prognosis (3). This emphasises the importance of establishing the diagnosis using the percutaneous technique rather than surgery which exposes the patient to unnecessary surgical morbidity.
For diffuse large B-cell lymphoma, the CHOP chemotherapy regimen (cyclophosphamide, doxorubicin, vincristine, and prednisone) is the conventional first-line treatment (25). The inclusion of rituximab to the CHOP regimen has recently proved to improve overall survival in individuals with primary adrenal lymphoma. With the addition of rituximab to the CHOP regimen, Kim et al. reported a two-year overall survival rate of 68.3 percent, with full remission attained in 54.8 percent of patients (26). The course of treatment usually requires about 6 cycle and each cycle lasts approximately 21 days. The usual dose of Rituximab is 375 mg/m2, Vincristine is 1.4 mg/m2, Doxorubicin is 50 mg/m2, and Cyclophosphamide is 750m g/m2. Unfortunately, the two represented patients in our study passes away within the first 2 - 3 months after surgery. Regarding the rest of the patients, four patients achieved full remission, seven patients died within the first year of treatment, three patients are still under treatment and the last patient had a relapse disease after 6 years of primary treatment.
The role of radiation therapy in the management of primary adrenal lymphoma is likewise unclear. Prolonged remission was reported in some patients with low-grade lymphoma and incomplete surgical excision treated with radiation therapy alone (27). However, more research into the role of radiation is required.
In patients with poor prognostic characteristics, CNS prophylaxis should be considered. This includes patients with high LDH level, advanced age and/or a high IPI score. In individuals with aggressive disease, CNS recurrence may be reduced by prophylactic intrathecal methotrexate and hydrocortisone injection; however, the findings are not yet verified (22).
The prognosis for PAL is bleak, with more than 90% of patients dying within one year after the initial diagnosis (28). An advanced age at diagnosis, a high tumour size, adrenal insufficiency at the time of presentation, and elevated LDH levels are all poor prognostic markers (29). Nine of the 17 patients in our study have passed away within the first year of diagnosis.
Generally, the current study has some limitation. First, it was a retrospective study, and thus we could not have access to all patients’ data. Second, the number of included patients was limited. Third, the outcomes in this study were not significantly different from other studies. On the other hand, the data included in the review regarding the diagnosis and management were up to date, and to our level of knowledge the number of primary adrenal lymphoma cases reported in the literature is less than 200 cases.
3.1. Conclusions
Primary adrenal lymphoma is a rare disease and the majority of our knowledge comes from rare cases reported in the literature. The disease is associated with poor outcomes. The role of surgery is limited and chemotherapy is the gold standard of treatment.
