





1. Context
Psychological stress is thought to be involved in the initiation (causation) and development of several pathological disorders (1) including type 1 and 2 diabetes mellitus (DM) (2, 3) and cancer (4). These disorders are considered the major causes of mortality worldwide (5, 6). The risk of developing cancer increases in individuals exposed to some genetic/environmental factors, such as psychological stress, radiation, obesity, tobacco use, alcohol consumption, and sunlight exposure, as well as genetic factors, such as genetic mutations and epigenetic alterations (5). The progression, tumor growth, and metastasis have been associated with stress, depression, anxiety, and other psychological abnormalities (5). Chronic stress can initiate psychological or physical damage by the abnormal secretion of stress-related mediators. These mediators can cause and even promote cancer by altering the genetic contents and regulations (5). A strong relationship has been shown between diabetes and psychological stress in the literature (7). DM, a chronic metabolic disorder, results from the combination of insulin resistance and insufficient insulin secretion from pancreatic β-cells (6, 8). In genetically susceptible individuals, type 1 DM is usually characterized by the autoimmune response to pancreatic β-cells. Possibly, these autoimmune processes are triggered by certain acquired/environmental factors, such as β-cell stress (β-cell stress hypothesis), quick somatic growth (the accelerator hypothesis), and viral infections and/or lower exposure to infectious agents in early childhood (the hygiene hypothesis) (9). The β-cell stress hypothesis suggests that various factors such as psychological stress can increase insulin demand, leading to β-cell stress (9). During recent years, psychological stress has been considered a risk factor in the progression of β-cell destruction (2). Recent evidence shows that inflammatory processes are associated with certain metabolic disorders such as metabolic syndrome, insulin resistance, and diabetes. It has been hypothesized that the inflammatory processes could be developed upon the exposure to chronic or acute psychological stress which is inducedby the stress hormones such as cortisol, epinephrine (EP), and norepinephrine (NE) or activates the renin-angiotensin system (RAS) (3). Therefore, the involvement of psychological stress in the onset of DM is an old hypothesis and seems to be undeniable (2).
2. Objectives
The aim of this review was to evaluate the molecular and cellular mechanisms by which psychological stress contributes to the initiation and progression of diabetes and cancer.
3. Evidence Acquisition
The following terms “psychological stress”, “diabetes”, and “cancer” were used for the search of research articles in some comprehensive databases namely, Web of Science, PubMed, Scopus, Embase, and Google Scholar. The literature search was limited to articles published up to September 2018. The current review paper focused on the studies related to the impacts of psychological stress on diabetes and cancer.
4. Results
4.1. Psychological Stress
The concept of stress was first introduced by Hans Selye, who is the father of studies dealing with the stress function in a medical context (10). In the biological field, he defines stress as a nonspecific and predictable response of the body to any demand (11). The stressors are subdivided into broad categories that include psychological (neurogenic or processive) or physiological (or physical) stressors. Psychological stress is primarily defined as an unpleasant emotional experience, usually accompanied by predictable behavioral, physiological, and biochemical responses (12). Physiologically, stress includes unpleasant phenomena that could cause some perturbations in the homeostasis of the human body as a result of the direct effect on the mind (12). Psychological stressors such as exams and/or video games (13) can influence biological and physiological systems (14-16). In other words, stress can affect the ability of the human body to resist to pathological conditions (17).
4.2. Molecular and Cellular Mechanisms Linking Psychological Stress to Diabetes
Psychological stress can cause various alterations in the levels of different biomarkers such as cortisol (18), testosterone (15), alpha-amylase (16), immunoglobulin A (IgA) (14), EP, NE, and glucagon, and it is also capable of activating RAS (3). These changes might play an essential role in the pathogenesis of a group of disorders. As depicted in Figure 1, psychological stress could give rise to an impairment in the HPA axis, SNS function, lipid profile, Th1/Th2 cytokine balance, RAS functionality, and insulin pathway. In the following sections, we discuss the molecular mechanisms underlying these events in detail.

Figure 1.
The effects of psychological stress on the development of diabetes through the HPA axis, SNS, lipid profile, inflammatory cytokines, RAS, and insulin resistance. Abbreviations: HPA, hypothalamic-pituitary-adrenal; IC, inflammatory cytokines; IR, insulin resistance; LP, lipid profile; RAS, renin-angiotensin system; SNS, sympathetic nerves system.
4.2.1. Effects of Psychological Stress on the Hypothalamic–Pituitary–Adrenal Axis and Sympathetic Nerves System
Chronic stress activates the HPA axis and increases the circulating levels of cortisol (18). The increased concentrations of cortisol may participate in the development of hypertension, hyperlipidemia, glucose intolerance, type 2 DM, cardiovascular diseases, neuropsychological disturbances, osteoporosis, cytokine-mediated immunologic responses, and abdominal obesity (18, 19).
Furthermore, there is an interdependence between the function of cortisol and catecholamines, considering that the cortisol receptors are localized on sympathetic nerves (20). Hence, the HPA axis stimulates the SNS activity which, in turn, increases the level of catecholamines (i.e., NE and EP) (21) thought to be involved in the induction of insulin resistance (22). Accordingly, patients with pheochromocytoma, in which large amounts of catecholamines are secreted, show a very low degree of insulin sensitivity (22). It has been implicated that NE incites the production of tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6), thereby leading to the β-adrenergic response. NE is also able to increase the sensitivity of adipocytes to TNF-α and IL-6 (23). Proinflammatory cytokines have been indicated to induce insulin resistance through the dysregulation in the insulin signaling pathway (23). In line with this, it has been shown that there is an attenuated response to psychological stress in mice deficient for IL-6 or TNF-α (23).
4.2.2. Effects of Psychological Stress on Lipid Profile
As mentioned earlier, chronic psychological stress promotes the secretion of glucocorticoids and catecholamines. These mediators can stimulate the release of excessive amounts of free fatty acids (FFAs) from visceral adipose tissue during the lipolysis process (24). On the other hand, an elevated level of other lipolytic factors such as glucagon, leptin, IL-6, and TNF-α has been reported during the psychological stress (25). FFAs induce the toll-like receptor 4 (TLR4) signaling pathway, stimulating the adipocytes to secrete inflammatory adipokines [monocyte chemoattractant protein-1 (MCP-1), IL-6, and TNF-α]. It has been shown that the unregulated secretion of adipokines can exacerbate the accumulation of monocytes, leading to the impairment of insulin sensitivity (24).
Accordingly, insulin suppresses hormone-sensitive lipase activity in adipocytes (25) as in the case of insulin resistance, as well as in the presence of excessive lipolytic factors, the activity of hormone-sensitive lipase would increase, causing the consecutive release of FFA and glycerol into the portal blood and liver (25). These events increase the rate of gluconeogenesis along with the secretion and synthesis of very low-density lipoproteins (VLDL) in the liver. The increased rate of gluconeogenesis leads to glucose intolerance (26). Besides, the accumulation of FFAs diminishes hepatic insulin binding through FFA oxidation, accompanied by a decrease in receptor-mediated degradation and uptake of insulin, which could result in the reduction of insulin clearance (hyperinsulinemia) (26). There is an inverse relationship between high-density lipoprotein (HDL) and the concentration of insulin. Regarding the anti-inflammatory properties of HDL, it would be plausible that hyperinsulinemia could activate the inflammatory responses (26). The accumulation of FFAs in muscle tissue can trigger insulin resistance, which is accompanied by the inhibition of insulin-induced glucose uptake and the decrease of glycogen synthesis in this tissue (27). FFAs also have lipotoxic effects on the pancreatic β-cells and therefore, decrease the insulin secretion. Taken together, these events contribute to hyperglycemia and glucose intolerance (27).
4.2.3. Effects of Psychological Stress on the Renin-Angiotensin System
The chronic psychological stressors have been demonstrated to increase the circulating levels of plasma angiotensin II and renin (28). Angiotensin II stimulates the secretion of MCP-1 through the angiotensin II type 1 receptor (AT1), activating the redox-sensitive and RhoA-dependent pathways, which could result in the accumulation of monocytes (29). Moreover, chronic exposure to stress can increase the expression of adipose angiotensinogen that activates adipogenesis and systemic RAS (30). The activation of adipose RAS inhibits the insulin signaling pathway, leading to inflammation in adipose tissue (31). A number of studies have indicated that the inhibition of angiotensin-converting enzyme (ACE) improves the insulin resistance (32) and the use of AT1 blockers can delay the onset of DM (33). Therefore, psychological stress-induced RAS activation increases the risk of DM.
Adipose-derived angiotensinogen is positively regulated by angiotensin II, insulin, SNS activation, and inflammatory adipokines such as IL-6 and TNF-α (30). Therefore, stress-induced insulin resistance can increase angiotensinogen (29). RAS, psychological stress, inflammatory mediators, and insulin resistance synergistically increase adipose tissue inflammation by positive feedback (30). Thus, the interplay among pathways mentioned earlier amplifies the strength of each pathway making individuals susceptible to develop DM.
4.2.4. Effects of Psychological Stress on Insulin Resistance
As mentioned above, the long-term exposure to stress can induce insulin resistance through several mechanisms. Chronic stress directly activates the innate immune system, which, in turn, activates the production of IL-6 and other cytokines associated with the acute phase response (34). Inflammatory cytokines play a crucial role in the induction of insulin resistance, and they could be considered as predicting markers for the development of DM (34). Pro-inflammatory cytokines, especially IL-6 and TNF-α, have been proposed to induce insulin resistance by the dysregulation of the insulin signaling pathway. These pro-inflammatory cytokines can phosphorylate the insulin receptor substrate-1-associated proteins along with the insulin receptor, resulting in the inhibition of the insulin binding and signaling (23). Correspondingly, acute mental stress induces peripheral insulin resistance that starts 60 minutes after the maximum stress (35). Therefore, the transient insulin resistance may be taken into account as a mechanism by which psychological stress increases the risk of type 2 DM (36). Insulin resistance might be considered a significant mechanism for the acquisition of type 2 DM and neurodegenerative diseases (37, 38). Hence, insulin resistance and psychological stress have synergistic effects on the emergence of DM (37, 38).
4.3. Molecular and Cellular Mechanisms Linking Psychological Stress to Cancer
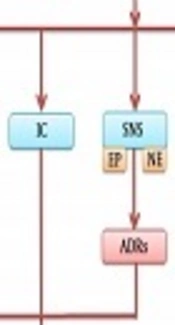
Numerous studies have been conducted to unravel the link between psychological stress and the initiation and progression of cancer at the molecular, biochemical, and systemic levels (39, 40). Psychological stress, as a result of chronic exposure to stress, is a major risk factor for the development of cancer, tumor growth, and metastasis (39, 40). The pathways involved in psychological stress and tumor cells are converged to some extent as they share some similar stress-induced mediators and receptors (39, 40). This convergence is responsible for the increased risk of cancer in individuals exposed to psychological stress. As shown in Figure 2, psychological stress can take part in the onset and progression of cancer through influencing the HPA axis, SNS, inflammatory cytokines, and the levels of oxytocin and dopamine. In the following sections, we evaluate the mechanisms linking the psychological stress to the initiation and progression of cancer.

Figure 2.
The effects of psychological stress on the development of cancer through the HPA axis, SNS, and inflammatory cytokines, as well as a decrease in the levels of oxytocin and dopamine. Abbreviations: ADRs, adrenergic receptors; DA, dopamine; EP, epinephrine; HPA, hypothalamic-pituitary-adrenal; IC, inflammatory cytokines; NE, norepinephrine; OT, oxytocin; SNS, sympathetic nerves system.
4.3.1. Psychological Stress-Induced Changes in Adrenergic Receptors and Their Association with Cancer
It has been established that during chronic stress, the concentration of catecholamines elevates in tumor tissues. The catecholamine receptor is differentially expressed in different types of tumors, such as pituitary adenoma, breast cancer, and prostate cancer (41). The adrenergic receptors (ADRs) play a vital role in catecholamine signaling pathways (42). ADRs are markedly expressed on cancer cells and in the tumor microenvironment such as adipocytes, macrophages, fibroblasts, and endothelial cells (42). When the ADRs signaling pathways are activated, they can alter the function and activity of tumor cells and the tumor microenvironment including the hindrance of apoptosis and DNA repair, as well as increased secretion of angiogenic factor (41). ADRs are the members of the G protein-coupled receptor superfamily and they are divided into two main groups including α-ADR and β-ADR (43). The α-ADR group is classified into α1 and α2 subtypes among which α1 couples to Gq. The G-protein can induce phospholipase C (PLC) and produce inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG) (44). The activated IP3 receptors, localized on the smooth endoplasmic reticulum (SER), increase the cytosolic levels of calcium. Furthermore, DAG and Ca2+ induce the activation of protein kinase C (PKC), an enzyme phosphorylating various molecules in various signaling pathways (44). On the other hand, α2 couples to Gi subtype and inhibits cyclic adenosine monophosphate (cAMP)-dependent protein kinase (PKA) by a reduction in the concentration of cAMP (45).
Activated β-ADR by Gs induces adenylyl cyclase (AC), an enzyme that activates PKA, thereby increasing the levels of cAMP within the cytosol that can stimulate the activation of PKA. The activated PKA is capable of changing the expression of various genes through regulating the activity of some transcription factors, such as the cAMP response element-binding protein (CREB), as well as the phosphorylation of downstream signaling molecules (42).
4.3.2. Stimulation of α-ADRs in Tumor Tissue
The stimulation of α-ADRs prevents apoptosis and enhances cell cycle progression due to proto-oncogenic effects on tumorigenesis (41). The α1-ADR signaling pathway induces the expression of some proto-oncogenes such as c-Jun and c-Fos (46, 47). The activation of α-ADR signaling contributes to the onset and progression of cancer as confirmed by in vitro and in vivo studies conducted on the growth of breast tumors (48, 49). The α-ADR signaling induces the activation of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/Akt pathway or mitogen-activated protein kinase (MAPK) signaling, and also increases the activator protein 1 (AP-1) binding affinity, thus promoting cellular survival. In addition, the activation of PLC through α-ADR can increase the synthesis of arachidonic acid involved in the cAMP/PKA pathway and the phosphorylation of CREB. The phosphorylated form of CREB in the cAMP/PKA pathway binds to the cyclin D promoter. Cyclin D controls the transition of the G1/S phase and stimulates the proliferation of the cells (48, 49).
4.3.3. Stimulation of α-ADRs in the Tumor Microenvironment
Chronic stress indirectly enhances the cancer progression and metastasis by the stimulation of the ADRs pathways in the tumor microenvironment (41). Angiogenesis and vasculogenesis supply oxygen and nutrients necessary for cancer progression (41). The activated α-ADR signaling pathway promotes vasculogenesis and angiogenesis in the endothelial cells of the tumor microenvironment (50). NE activates α-ADR that induces the migration and proliferation of the endothelial progenitor cells (EPCs) through the activation of the PI3K/Akt/endothelial NOS (eNOS) pathway (50). Furthermore, NE elevates the number of EPCs, as well as the concentration of vascular endothelial growth factor (VEGF) in blood circulation (50).
Macrophages play a major role in chronic inflammation, and the presence of these cells promotes the progression of cancer. These cells contribute to the processes of angiogenesis and cell proliferation, thereby releasing different cytokines including TNF-α, IL-6, and VEGF (51). NE activates the α2-ADR signaling pathway in macrophages and stimulates lipopolysaccharides (LPS)-induced production of TNF-α through the MAPK pathway (51). Additionally, the extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 (belonging to the MAPKs) are phosphorylated upon the NE induction (51). Fibroblast cells play a significant role in the stimulation of tumor growth through the release of growth factors such as stromal cell-derived factor-1 (SDF-1). In addition, fibroblast cells enhance the angiogenesis process by the recruitment of EPCs (52). The activated α2-ADRs expressing on fibroblast cells enhance the proliferation and growth of these cells. The increased proliferation of fibroblasts increases the levels of growth factors in the tumor microenvironment (53).
4.3.4. Stimulation of β-ADRs in Tumor Tissue and Tumor Microenvironment Cells
The β-ADR signaling pathway induces the activity of AC enzyme, mediated by the Gs protein-coupled β2-adrenergic receptor in which AC enzyme converts ATP to cAMP that, in turn, induces the activation of the cAMP/PKA signaling pathway. This pathway regulates the expression of genes involved in the tumor progression such as VEGF, IL-8, IL-6, and matrix metalloproteinase (MMP). In addition, this signal stimulates the DNA damage (41) by the induction of some transcription factors, such as CREB, NF-κB, and AP-1, in various types of cancers (54). In stress conditions, the cAMP/PKA signaling pathway is activated in tumor cells and then it elevates the affinity of AP-1 and CREB to bind the gene promoters of IL-8 and IL-6 (55). The cytokines lead to enhanced tumor growth, angiogenesis, and metastasis in tumor cells and tumor microenvironment. It has been implicated that the increased level of IL-6 increases the invasiveness of melanoma cells (55). Furthermore, catecholamines in ovarian cancer cells induce the expression of IL-6 and enhance its secretion through the activation of Src. NE stimulates the phosphorylation of Src through the cAMP/PKA pathway (56, 57). In addition, β-ADR-induced Src activation improves the kinase activity of the focal adhesion kinase (FAK) protein in which the tyrosine at residue 397 is phosphorylated. This kinase governs cell migration, which is needed for the adherence of tumor cells to the extracellular matrix (ECM). FAK suppresses anoikis (a form of programmed cell death) caused by the inappropriate cell-ECM connection (58). Therefore, this signal protects cancer cells from anoikis (59). The EP-induced PKA activation leads to the phosphorylation of the serine at residue 211 in the Bcl-2-associated death promoter (BAD) gene. When the BAD protein undergoes phosphorylation, it is inactivated and its interaction with Bcl-xL and Bcl-2 (proteins preventing cytochrome c-induced cell death) is decreased (60). Finally, this pathway enhances the anti-apoptotic effects of Bcl-xL and Bcl-2, leading to the inhibition of apoptosis (41). Jagged 1 protein is a membrane factor that acts as a ligand for the interaction with the Notch receptors, expressed on neighboring cells. The interaction of Jagged 1 protein and Notch receptors influences the apoptosis and cell proliferation (41). The NE-induced Jagged1/Notch signaling pathway enhances tumor angiogenesis and stimulates the Jagged 1 expression in breast tumor (61). The mammalian target of rapamycin (mTOR)/p70S6K pathway is a predominant pathway that mediates the NE-stimulated expression of Jagged 1 (61). The activation of the β-ADR signaling pathway stimulates the DNA damage via the β-arrestin-1 signaling cascade, leading to an increased risk of tumor initiation (62). β-arrestin activates the PI3K/Akt pathway, leading to the phosphorylation of E3 ubiquitin ligase murine double minute 2 (MDM2). The phosphorylated form of MDM2 inhibits DNA repair by the degradation of the p53 protein (62). The engagement of NE in β2-ADR activates PLC through the β-arrestin/Src cascade. The activated form of PLC enzyme catalyzes PIP2 to produces IP3 and DAG. Upon the production of IP3, the cytosolic content of Ca2+ is elevated and the PKC activation is stimulated, leading to the enhanced migration of cancer cells (63). Catecholamines are capable of elevating the expression of MMP-2/9, as well as the invasiveness and metastasis of ovarian tumor cells, leading to the PKA-induced phosphorylation of STAT3 transcriptional factor. The phosphorylated form of STAT3 regulates the MMPs expression (39). In addition, β-ADR can initiate the growth of the pancreatic tumor by means of the ERK/MAPK signaling pathway, which regulates the transcription of VEGF and MMP-2/9 (41). In a study by Pan et al., they indicated that in hemangioma cells, NE decreases the expression of p27 and p21 and increases the expression of the cyclin D2 and A2, leading to cell cycle progression (64). NE stimulates the expression of VEGF, NO, and MMP-9, which are all regulated by the PI3K/Akt/eNOS signaling pathway. It has been shown that VEGF, NO, and MMP-9 are associated with the tube formation and invasiveness of hemangioma cells (64). The β2-ADR activates the expression of the NF-κB protein, which can up-regulate the expression of both PTGS2 and PTGES (enzymes involved in the synthesis of prostaglandin E2). Prostaglandin E2 is a releasing factor involved in cancer metastasis that plays a central role in inflammatory processes (65). In breast cancer cells, the activated β2-ADR up-regulates the expression of human epidermal growth factor receptor 2 (HER2) through the activation of STAT3. The upregulation of HER2 elevates the expression of β2-ADR, thereby activating the ERK signaling pathway. Therefore, two pathways can amplify each other and promote the effects of stress on cancer progression (66). In the tumor microenvironment, the proliferation of macrophages is promoted by the β1-ADR/cAMP/PKA pathway, leading to the increased production of TNF-α (41). TNF-α suppresses the immune response against tumors, stimulates tumor angiogenesis, and reconstructs the stromal tissue to enhance tumor progression (67). In addition, NE stimulates the ability of pro-angiogenic of EPCs through the β2-ADR pathway (50).
4.3.5. Psychological Stress-Induced HPA Axis Response in Cancer
Glucocorticoids boost the growth of tumor cells through the inhibition of the MAPK pathway and protect those cells from apoptosis (68). The activated glucocorticoid receptors blunt the MAPK signaling pathway via the stimulation of the MAPK phosphatase-1 (MKP-1) expression (68). Increased MKP-1 promotes tumor growth and prevents cell death. The MAPK signaling pathway plays an essential role in the induction of apoptosis. MKP-1 inhibits the activation of the JNK and ERK1/2 pathways, thereby reducing the phosphorylation rate of the Ets-like transcription factor-1 (ELK-1) transcription factor, as well as enhancing the cell survival (68).
4.3.6. Effects of Psychological Stress on Dopamine and Oxytocin
Dopamine, a precursor of EP and NE, is produced within the CNS (69). Unlike EP and NE, the amount of dopamine is decreased during chronic stress. Dopamine inhibits the cancer progression and blocks VEGF-mediated angiogenesis and tumor cell proliferation (70). In addition, dopamine and the agonist of the dopamine D2 receptor (D2R) decrease the process of angiogenesis and cell growth of lung cancer cells (71). Oxytocin hormone modulates the HPA axis and decreases stressful feelings. Similar to dopamine, oxytocin is reduced in chronic psychological stress (41).
5. Conclusions
In the present study, we summarized the association of psychological stress with the risk of diabetes, as well as the initiation and development of cancer. Based on our findings, it seems that psychological stress plays an axial role in the initiation and progression of diabetes and cancer. Moreover, psychological stress may induce the incidence or progression of diabetes through influencing the HPA axis, SNS, lipid profile, inflammatory cytokine levels, RAS, and the insulin signaling pathway. Of note, psychological stress may activate cancer cells, as well as the surrounding cells in the tumor microenvironment through the HPA axis, SNS, and inflammatory cytokines, along with reducing the levels of oxytocin and dopamine. Based on the above statements, the role of psychological stress is undeniable in the onset and progression of diabetes and cancer. The identification of the pathways involved in this scenario would open up a new horizon to understand how emotional changes predispose individuals to develop cancer and diabetes and help the scientists to find master key genes to fight these disorders.