




1. Background
Infantile nephrotic syndrome (INS) is defined as nephrotic syndrome (NS) that refers to disease being present after the first three months of life up to one year of age (1). About 90% of patients are steroid responsive and approximately one third of the remaining 10% do not respond to corticosteroids; the so called steroid resistant nephrotic syndrome (SRNS). It progresses to end-stage renal disease (ESRD) after about 10 years (2-6). It is well known that inherited structural defects of the glomerular filtration barrier are responsible for a large proportion of SRNS cases. Glomerular filtration barrier structurally consists of glomerular basement membrane (GBM), endothelial cells and podocytes. There are slit diaphragms between foot processes of podocytes. Main structural elements of the slit diaphragm are nephrin, podocin and CD2AP encoded by NPHS1, NPHS2 and CD2AP genes, respectively (7). Indeed, mutations in genes highly expressed in podocytes have been found in two thirds of patients presenting with SRNS in the first year of life (8-10).
2. Objectives
In this study, we aimed to analyze genotypic and phenotypic features of both NPHS1 and NPHS2 genes in INS and prognostic effect of E117K polymorphism in NPHS1 gene as E117K change was accepted as polymorphism of NPHS1 gene.
3. Methods
3.1. Study Design, Inclusion and Exclusion Criteria and Data Collection
The study was enrolled retrospectively, with forty eight infantile NS cases diagnosed and followed in our clinic. An informed consent was obtained after a complete explanation for the patients’ parents. The institutional committee has approved the study. Children with syndromic NS, secondary NS and having irregular follow-up or missing data on their records were excluded from the study. Demographic features, clinical data, genetic analysis and outcomes were extracted from their charts.
The diagnosis of infantile NS was done by the presence of severe proteinuria (> 40 mg/m2/hour or urine protein/urine creatinine ratio of > 2 mg/mg), edema, hypoalbuminemia (< 2.5 mg/dL), and hyperlipidemia. A complete response was defined as both clinical healing and disappearance of proteinuria over three consecutive days. Presence of proteinuria below nephrotic range without edema and hypoalbuminemia was defined as partial response. Failure of remission after 4 weeks of prednisone treatment indicates steroid resistance (11). The glomerular filtration rate (GFR) was measured according to guidelines published by K/DOQI (12).
Patients were classified into three groups; group 1: cases having only NPHS1 mutation; group 2: cases with only NPHS2 mutation; group 3: cases with any mutation. Patients having E117K polymorphism in NPHS1 gene were separately compared with each of the 3 groups.
3.2. Mutational Analysis of NPHS1 and NPHS2
Peripheral blood leukocytes were used for genomic DNA extractions from patients and healthy controls by using Purelink Genomic DNA Mini Isolation Kits (Invitrogen, Carlsbad, CA). Thermo Scientific Nanodrop spectrophotometer (Wilmington, USA) was used for quantification extracted DNA purity at 260/280 nm. For quality assessment, 2% agarose gel electrophoresis was used. The direct sequencing of all 29 exons of NPHS1 gene and 8 exons of the NPHS2 gene were made. The direct DNA sequencing reactions were performed from controlled DNA of NPHS1 and NPHS2 genes. Nucleotide comparison of the NPHS1 cDNA sequence (NCBI reference sequence: NM_004646) and NPHS2 cDNA sequence (NCBI reference sequence: NM_014625) were performed using GeneMapper SeaScape Software v3.0. We carried out amino-acid comparisons with NCBI reference sequence: NP_004637 protein database for NPHS1 gene and NCBI reference sequence: NP_055440 for NPHS2 gene protein database.
3.3. Statistical Analysis
Results were shown as mean ± SD. For the comparisons between two continuous variables t-test was used. Genetic association between NPHS1 variants and NS were analyzed by chi-square test or Fisher’s exact test. The difference was considered to be significant if P < 0.05. Patients having NPHS1 mutation were compared with other patients. Statistical analyses were performed using SNPStats software.
4. Results
4.1. Baseline Characteristics
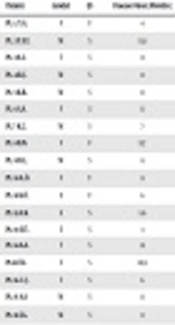
Forty-eight children with infantile NS (18 girls) were included in the study. The mean age of onset was 8.7 ± 2.3 months and the follow-up time was 8.3 years. Consanguinity was seen in 12.5%. Seven familial and 41 sporadic cases were included in the study. Kidney biopsy was carried out in 45 patients; there were focal segmental glomerulosclerosis in 29 (65%), IgM nephropathy (Ig MN) in 6 (13%), and minimal change disease (MCD) in 10 (22%) (Table 1).
| Patient | Gender | F/S | Disease Onset (Months) | Treatment/Response | Biopsy | CKD/When? (Months) | Tx/Age (Months) | NPHS1 | NPHS2 |
|---|---|---|---|---|---|---|---|---|---|
| Pt. 1. Y.G. | F | F | 4 | ARB,ACEI/- | FSGS | ESRD/11 | +/28 | V709G het. | |
| Pt. 2 Y.E.T. | M | S | 11.8 | CS/+ | FSGS | - | - | R408Q het. | - |
| Pt. 3 A.S. | F | S | 8 | CS/+ | FSGS | - | - | R800C het. | - |
| Pt. 4 R.Ç. | M | S | 11 | CS + | FSGS | N1077S het. | |||
| Pt. 5 K.B. | M | S | 11 | CS,CyC,CsA,ritux/-,-,-, | IgM | - | - | N1077S het. | - |
| Pt. 6 S.A. | F | S | 11 | CS,CyC,CsA/-,-,+ | FSGS | - | - | - | V180M homo. |
| Pt. 7 K.Ç. | M | S | 7 | CS,CyC,CsA/-,+,+ | FSGS | Stage 2/180 | - | - | P89T het. |
| Pt. 8 B.N. | F | F | 11.7 | CS,CyC,CsA/-,+,+ | FSGS | - | - | - | P89T het. |
| Pt. 9 D.Ç. | M | S | 8 | CS,CyC,CsA/-,-,- | MCN | ESRD/ 90 | +/121 | - | 467 ins. 7 T homo. |
| Pt. 10 A.Ö. | F | F | 8 | FSGS | ESRD/22 | +/60 | - | P20L/R168H comp het. | |
| Pt. 11 K.Ö. | E | F | 6 | ESRD 5/11 | +/96 | - | P20L/R168H comp het. | ||
| Pt. 12 A.K. | E | S | 5.6 | FSGS | P118L homo. | ||||
| Pt. 13 E.Ü. | E | S | 3 | MCN | P20L homo. | ||||
| Pt. 14 A.A. | F | S | 12 | CS,CsA/-,- | FSGS | P20L/R168H comp het. | |||
| Pt.15 Ö.I. | F | S | 10.9 | CS/CyC/-- | FSGS | ESRD/ 25 | +/76/reject | P20 L homo. | |
| Pt. 16 C.Ç. | F | S | 6 | CS,CyC/-,- | FSGS | Stage 4/9 | - | E117K het. | P118L het. |
| Pt. 17 A.A. | M | S | 11 | CS,CyC,CsA /-,-,+ | MCN | - | - | E117K het. | V64E/K289X het. |
| Pt. 18 İ.G. | M | S | 9 | CS,CyC,CsA/-,+,+ | FSGS | E117K het. | R229Q het. | ||
| Pt. 19 D.P. | F | S | 6 | CS,CyC,CsA,CQ10/-,-,+,+ | FSGS | Stage 2/13 | |||
| Pt. 20 S.Ç. | M | S | 12 | CS,CyC,CsA,CQ10/-,-,+,+ | FSGS | ||||
| Pt. 21 M.B. | M | S | 9 | CS,CyC/-,- | FSGS | ESRD 5/12 | +/20 | - | |
| Pt. 22 S.K. | M | S | 11 | CS,CsA,CQ10/-,+,+ | FSGS | Stage 2/17 | - | - | |
| Pt. 23 S.A. | M | S | FSGS | ||||||
| Pt. 24 Y.D. | M | F | 6 | ARB/ACEI | FSGS | ESRD/18 | +/26 | - | |
| Pt. 25 S.G. | F | S | 12 | CS,CyC/+,+ | IgM | - | - | - | |
| Pt. 26 A.A. | M | S | 10.8 | CS,CyC,CsA/-,+,+ | IgM | - | - | - | |
| Pt. 27 H.Ç. | M | S | 12 | CS/+ | MCN | - | - | ||
| Pt. 28 F.A. | F | S | 11 | CS/+ | |||||
| Pt. 29 B.Ç. | M | S | 10.5 | CS,CyA/-,+ | FSGS | - | - | - | |
| Pt. 30 E.A. | M | S | 11 | CS,CyC,CsA/-,+,+ | FSGS | ||||
| Pt. 31 H.Ç. | F | S | 12 | CS/+ | MCN | ||||
| Pt. 32 B.N. | F | F | 4 | CS,CyC,CsA/-,+,+ | FSGS | ESRD/20 | +/60 | ||
| Pt. 33 C.D. | F | S | 11 | FSGS | |||||
| Pt. 34 M.B. | M | S | 11.3 | CS+ | MCN | - | |||
| Pt. 35 M.E. | M | F | 11 | CS/+ | FSGS | ||||
| Pt. 36 A.K. | F | S | 12 | CS,CyC,CsA/-,-,+ | ESRD/21 | +/100 | - | ||
| Pt. 37 M.P. | M | S | 11 | FSGS | ESRD/35 | +/122 | |||
| Pt. 38 O.D. | M | S | 10 | IgM | |||||
| Pt. 39 M.A. | M | S | 9 | CS,CyC,CsA,ritux/-,-,?,-, | FSGS | Stage 3/144 | - | E117K het. | - |
| Pt. 40 A.H. | M | S | 7 | CS/+ | MCN | - | - | E117K het. | - |
| Pt. 41 R.K. | M | S | 6.1 | CS/+ | FSGS | - | - | E117K het. | - |
| Pt. 42 Ö.Y. | M | S | 7.2 | CS,CyC/+-,+ | MCN | - | - | E117K het. | - |
| Pt. 43 G.Ç. | F | S | 5.7 | CS,CsA/-,+ | IgM | - | - | E117K het. | - |
| Pt. 44 D.A. | F | S | 3 | CS,CsA/-,+ | MCN | - | - | E117K het. | - |
| Pt. 45 E.Y. | M | S | 5.1 | CS,CyC/-,+ | MCN | - | - | E117K het. | - |
| Pt. 46 A.B. | M | S | 4 | ARB/ACEI | FSGS | Stage 4/10 | - | E117K het. | - |
| Pt. 47 A.Y. | M | S | 9 | CS,CyC/+,+ | IgM | - | - | E117K het. | - |
| Pt. 48 K.A. | F | S | 6 | CS,CyC/+,+ | FSGS | - | - | E117K het. |
Abbreviations: ARB, angiotensin receptor blocker; CKD, chronic kidney disease; CS, corticosteroid; CyA, cyclosporine A; CyC, cyclophosphamide; ESRD, end stage renal disease; FSGS, focal segmental glomerulosclerosis; GFR, glomerular filtration rate; het, heterozygous; homo, homozygous; IgMN, IgM nephropathy; MCD, minimal change disease; NS, nephrotic syndrome; PCR, polymerase chain reaction; Tx, transplantation.
Mutation analysis of NPHS1 and NPHS2 was performed in all cases, with a mutation rate of 58.3% (28 out of 48). Different DNA sequence variant analyses were performed. Missense, nonsense, insertions and deletions and splice site mutations resulting in nucleotide changes were expressed either as homozygous or compound heterozygous sequence changes.
Initially, all patients had normal renal function except one (patient 11). Patients were given corticosteroids, cyclosporine, cyclophosphamide, and rituximab. At the end of the follow-up period (8.3 years), there were 32 children with partial (n = 20) and complete response (n = 12) and 16 without any response. Among the 16 non-responsive cases, 6 developed chronic kidney disease (CKD) stages of 2 to 4, and 10 progressed to ESRD (Table 2).
| Variables | Patients with NPHS1 Mutation (Group 1) | Patients with NPHS2 Mutation (Group 2) | Patients Without Any Mutation (Group 3) | |||
|---|---|---|---|---|---|---|
| No E117K | With E117K | With E117K | No E117K | |||
| Patient number | 5 | 10 | 3 | 6 | 4 | 20 |
| Gender, male/female | 3/2 | 5/5 | 2/1 | 4/2 | 2/2 | 13/7 |
| Age of disease onset, month | 7.9 ± 3.9 | 8.32 ± 3.01 | 8.6 ± 2.5 | 6.2 ± 1.8 | 6.3 ± 0.8 | 9.9 ± 2.4 |
| Number of relapses | 4/patient | 6.2/patient | 4.3/patient | 6.1/patient | 3.8/patient | 7.1/patient |
| Response to treatment, complete/partial/no | 3/-/2 | -/3/7 | -/2/1 | 2/3/1 | -/3/1 | 7/9/4 |
| Proteinuria, mg/m2/h/ Cr. Cle. mL/min/m2 | 51.7 ± 14.3/ 67.1 ± 29.0 | 67.2 ± 21/ 32.3 ± 16 | 37.1 ± 9.7/ 35.8 ± 11.2 | 12.3 ± 6.1/ 86 ± 9.2 | 43.2 ± 11.9/ 87.1 ± 12.1 | 128.3 ± 22.9/ 22.3 ± 27.1 |
| CKD | - | 1 | 1 | 1 | 1 | 2 |
| ESRD | 1 | 4 | - | - | - | 5 |
Abbreviations: CKD, chronic kidney disease; ESRD, end stage renal disease.
Among the ESRD cases (n = 10) there was only one case with NPHS1 (V709G) mutation. Half of the ESRD cases had no mutation, though 40% revealed NPHS2 mutation (P20L, R168H, 467/7 insertion T). The median time for progression to ESRD was 36.1 ± 51.9 months. All 10 ESRD cases were transplanted at a median age of 68.9 ± 43.6 months, with only one rejection due to recurrence of the disease (Table 2). The relationship between histological diagnosis, age of onset, familial or sporadic form, clinical course, and mutational analysis are shown in Table 1.
4.2. Genotype-Phenotype Correlations of Groups
4.2.1. Group 1: Cases with Only NPHS1 Mutation (Patients 1 - 5)
There were 5 patients, three of them male, and three familial, with the mean disease onset of 7.19 ± 2.73 months. Patients 1 - 3 were heterozygotes for V709G, R408Q, R800C, and patients 4 - 5 were both heterozygotes for N1077S.
Kidney biopsy showed focal segmental glomerulosclerosis in four and immunoglobulin M nephropathy in one patient. There were two cases with chronic kidney disease (CKD); one progressed to stage 4 CKD in 10 months and had both E117K and N1077S. The second one, who progressed to ESRD within 11 months of disease onset and was transplanted with a well-functioning graft from her mother for four years, had V709G mutation on the 16th exon (patient 1) (Table 1). Thus, ESRD ratio was 20% (n = 1) in the group.
4.2.2. Group 2: Cases with Only NPHS2 Mutation (Patients 6 - 18)
There were 13 patients, eight of them male, and three familial cases with the mean disease onset of 8.3 ± 3.0 months in this group. Five cases (pt. 6, 9, 12, 13, 15) were homozygous, two heterozygous (pt. 7, 8), and three compound heterozygous (pt. 10, 11, 14). The most common mutation was P20L found in five cases; 3 of 5 had compound mutation with R168H (pt. 10, 11, 14). Pt. number 16, carrying both E117K and P118L, went to stage 4 CKD within 9 months, whereas the others were partially responsive to treatment (Table 1).
As patient number 11 was admitted in ESRD status, no biopsy was taken. Among the remaining nine cases, nine samples had FSGS. Four of them progressed to ESRD at a mean age of 37 ± 35.8 months. Patient number 10 and patient number 11 were siblings, having P20L and R168H compound heterozygous mutations, progressed to ESRD. All three patients had E117K single nucleotide polymorphism of NPHS1, accompanied with various NPHS2 mutations. Two other patients with ESRD were patients 9 and 15, with mutations of 467/7 insertion homozygous and P20L homozygous, respectively. All of the ESRD cases were transplanted and followed with a well-functioning graft, except for one rejection at the 76th month due to recurrence of the disease. There were only three patients that had partial response, having mutations of V180M homozygous, P89T heterozygous, and TT/AA 951T > C. A case with P118L homozygous, another case with P20L, and a third case from the compound mutated cases (P20L and R168H) were also not responsive to treatment (Table 1). Thus, ESRD ratio was 30.8% (n = 4) in the group.
4.2.3. Group 3: Cases Without any Mutation (19 - 48)
There were 30 patients, with a male: female ratio of 19:11, with three familial cases, and the mean onset of disease was 9.9 ± 2.4 months.
There were cases having renal biopsy, FSGS, IgM nephropathy, and MCD. Two cases progressed to stage 2 CKD within 13 and 17 months, whereas five patients progressed to ESRD and were transplanted with well-functioning grafts.
Pathogenic mutations in NPHS1 were found in 5/48 (10.4%) of cases. Also, the ratio of NPHS2-detected patients was 13/48. There were four NPHS2 mutated cases that progressed to ESRD (4/10; 40%). Whereas only one out of five NPHS1 mutated patients progressed to ESRD, this equals to 20% of cases. All 10 cases with E117K were sporadic, resistant to treatment, and had earlier onset (Tables 1 and 2).Thus, ESRD ratio was 16.7% (n = 5) in the group.
5. Discussion
NPHS1 gene mutations are the cause of the Finnish type nephrotic syndrome. In Finland, two mutations, Fin major and Fin minor were seen in 78% and 16% of the cases respectively (13). NPHS1 gene mutations account for 39% - 55% cases of childhood NS in European, North American, and Turkish societies and nearly 40% of CNS cases (14, 15). Nephrin encoding of mutations of NPHS1 are responsible for the most of congenital NS and result in infantile and childhood steroid-resistant nephrotic syndrome (SRNS) (16-19). Also, there are some infantile NS cases having both nephrin and podocin mutations, causing triallelic abnormality (homozygous mutations in one gene and heterozygous mutations in the other) (20). Because of INS genetic heterogeneity there is no clear genotype-phenotype correlations. A few patients with typical INS cases were found to lack NPHS1 mutations but were found to have recessive NPHS2 mutations (20). Philippe et al. reported NPHS1 mutations in 7% - 14% of the patients with SRNS at least 3 months after birth (age at onset of NS in mutated patients 0.5 - 8 years (mean 3 years)) (19). Santin et al. also supported the former with same mutation ratio with the age of onset of 0.7 - 27 years (mean 8 years) (21). Lahdenkari et al. pointed to the immunogenic stimuli caused by hypomorphic mutation in cases with NPHS1 variants (22). Also, infections trigger the attacks, Kitamura et al. reported two siblings bearing nephrin mutations with spontaneous partial remissions, but repeated relapses concurrently with respiratory infections. One of them presented NS at birth and the other at 10 months of age. Both were compound heterozygous for the p.C265R and p.V822M mutations. The p.C625R mutant protein was predominantly trapped within the ER, while the p.V822M protein reached the plasma membrane, explaining the milder phenotype (23). Another factor is environmental influences that play a role in phenotype variability. Santin et al. followed an adult case without any renal impairment during 2 years, diagnosed as FSGS at 27 years of age, with two NPHS1 mutations (p.R827X and p.R976S) (21). On the other hand, Philippe et al. identified one patient with infantile-onset NS with the same two mutations (19).
NPHS2 gene mutations account for an autosomal recessive steroid resistant nephrotic syndrome (SRNS) with early disease onset and focal segmental glomerulosclerosis (FSGS) (24). Infantile nephrotic syndrome cases can be derived from either isolated nephrin or podocin mutations, or both (25). There are other genes that cause INS as LAMB2 or WT. In this study, we evaluated both NPHS1 and NPHS2 mutations in the infantile group to determine nephrin and podocin mutational profile and outcome. A recent study reported 57% rate of mutations in INS, with 14% NPHS1, 29% NPHS2, and 14% WT1 (26). Our total mutation rate was 37.5%. Mbarek et al. reported ten different pathological mutations including NPHS1 and NPHS2 in 24 Tunisian children, and only two infantile cases without any NPHS1 or NPHS2 mutation (27). In our cohort, cases without mutations made up 62.5% (30/48) of our population, which might be explained so that we have only analyzed NPHS1, NPHS2 genes of INS.
Santin et al. analyzed podocyte genes in SRNS, with patient ages ranging from congenital to adult onset (21) Infantile group had various podocyte mutations with a rate of 57%. Also, NPHS1 mutation rate was found to be 14% and NPHS2 rate was 29% only in the infantile onsets. The range of NPHS1 gene mutations prevalence was 39 to 55% and the NPHS2 gene mutations was 10 to 28% in European and American populations (11, 12, 25, 28). In contrast to these, Abid et al. reported that the NPHS1 gene mutations were approximately 20% and NPHS2 gene mutations 5.5% of the patients with early onset NS (29). The prevalence of NPHS1 and NPHS2 were low in studies from Japan and China (30, 31). Our study group consisted of only infantile cases. We found the mutation rate to be 32%, nearly equal to the study reported by Santin et al. (21). Also, our NPHS2 rate was similar (20.8%). Our cohort was more relevant to European and American than Asian populations (11, 12, 19, 25, 30, 31). It shows geographic and ethnic genetic diversity of NS in the world.
Nowadays, papers have reported general non-responsiveness to intensive immunosuppressive therapy regimens, and many studies observed the low recurrence rate of NS after transplantation (22, 29, 32, 33). Similarly, our cohort showed a 16% rate of intensive immunosuppressive drug response and only one recurrence of disease after transplantation.
Tryggvason et al. reported the faster progression to ESRD of NPHS1 mutations compared to NPHS2 (34). We found a worse prognosis in the NPHS2 positive group; the difference is due to the population group, as the majority of NPHS1 positive cases in their study were congenital nephrotic syndrome cases, not infantile ones (34). NPHS1 gene mutations progressed rapidly to ESRD within one to three years of age in children in some studies (1, 33, 35). In addition, Abid et al. reported that NPHS1 gene mutations carriage in children result in preserving renal function up to 2.5 years of age (29). We had only one patient who progressed to ESRD within 11 months of disease onset.
Koziell et al. reported digenic inheritance of NPHS1 and NPHS2 genes (31). In the study of a cohort from Pakistan, they observed a patient with both heterozygous R408Q NPHS1 gene mutation and a heterozygous P321S mutation in the NPHS2 gene together (30). We had no digenic cases, but three of our cases had heterozygous E117K polymorphism in NPHS1 and P118L in NPHS2 in one, K289X in another, and R229Q in the third case.
Today, there are more than 173 various mutations of NPHS1 reported in the Human Gene Mutation Database. The clinical course associated with NPHS1 mutations is not restricted to classical CNS. NPHS1 mutations were causing a mild disease in adulthood onset NS with the FSGS histology (28). We can explain this by underlying mutations; predominately missense mutations result in minor protein modifications.
Abid et al. screened mutations of 145 patients, including 36 early-onset NS cases (CNS cases included). Mutations in the NPHS1 gene accounted for approximately 20% of cases with early-onset NS. They showed a heterozygous mutation, R408Q, in three patients with childhood onset (29). Lenkkeri et al. reported this mutation as a compound heterozygous condition in CNS cases (15). In our cohort, we found one patient with R408Q with disease onset of 11.8 months. Other NPHS1 mutations of our cohort were N1077S, V709G, and R800C.
Here, we also would like to discuss the phenotypic features of patients with the E117K genotype, which has been accepted as a polymorphism since Lenkkeri et al. reported it as a single nucleotide polymorphism (15). As our cohort did not deal with CNS cases, we questioned whether it is also a polymorphism in the infantile group or if it affects the protein as a hot mutation. E117K was found in 6 homozygous and 21 heterozygous conditions in the study by Abid et al. (29). However, this was a common variant, as it was found in normal individuals (1). In our cohort, E117K cases showed a clinical course that had no statistical differences compared with other nephrin mutations, whereas, E117K had statistically significant differences compared with non-mutated cases (Table 3). Also, we had previously reported patient number 26 in a case report, when she was in stage 2 CKD, and emphasized that, if E117K change in nephrin diverges the podocyte signaling pathways and causes P118L mutation of NPHS2, it behaves different and suggested that it might be called a genetic modifier in future (36). Pettersson-Fernholm et al. examined diabetic patients with nephrin polymorphism (37) and found that the onset of diabetes in patients with E117K polymorphism K genotype occurred later compared to patients with the wild genotype. Downregulation of the nephrin gene was seen in experimental nephrosis of rats. Animal models showed that the expression levels of nephrin-specific mRNA were associated with early changes of diabetic nephropathy (35). In the literature, it was reported that any changes in amino acid sequence might affect the nephrin protein confirmation. Even if the polymorphisms have unknown functional implications, they may have a role in proteinuria via influence on the slit diaphragm permeability (1, 36).
| Group | Patients with E117K (N:10) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Disease Onset (Months) | Familial / Sporadic | Histology (FSGS/Others) | ESRD | CKD | |||||||
| SD² | t | P | χ2 | P | χ2 | P | χ2 | P | χ2 | P | |
| Group 1, n = 5 | 5.72 | 0.91 | > 0.1 | 3.65 | > 0.05 | 3.36 | > 0.05 | 3.64 | > 0.05 | 0.75 | > 0.2 |
| Group 2, n = 13 | 6.3 | 1.87 | < 0.05 | 3.52 | > 0.05 | 1.82 | > 0.1 | 5 | < 0.05 | 0.39 | > 0.2 |
| Group 3, n = 30 | 6.41 | 3.47 | < 0.05 | 3.34 | > 0.1 | 1.68 | > 0.1 | 3.34 | >0.05 | 0.68 | > 0.2 |
Abbreviations: CKD, chronic kidney disease; ESRD, end stage renal disease; FSGS, focal segmental glomerulosclerosis.
Nephrin is a transmembrane adhesion protein (10) and directly participates in slit diaphragm structure by its ability to homo- or hetero-dimerization (1). NPHS1 missense mutations result in abnormal endoplasmic reticulum nephrin retention, and failure of trafficking out to the cell surface (20). Therefore, nephrin dysfunction may explain severe and early onset phenotypes resulted from mostly truncated and missense NPHS1 mutations. Beside these, extracellular Ig domains 2, 4 and 7 have clusters of mutations. More than 50% of missense mutations are extracellular, and 66% of them are in Ig domains and this leads to the hotspot mutations. The structure of nephrin is highly flexible, most mutations can affect it. E117K polymorphism is a missense mutation of nephrin and placed in immunoglobulin motif and the transmembrane domain of the polypeptide chain (22). Koziell et al. showed that missense NPHS1 mutation decreases nephrin expression in podocyte cell cultures (31).
Pettersson-Fernholm et al. reported that all the polymorphisms of E117K, N1077S, and R408Q were changing the amino acid (37). Any change in amino acid sequence such as G > A substitution in E117K is called a mutation, whether it leads to the protein malfunctioning (hot/disease-causing mutation) or not (polymorphism). In our patients having E117K polymorphism, there were statistically significant differences of disease onset when compared with other mutations. E117K had a similar phenotype to other known mutations of nephrin and podocin and differed from the non-mutated group (38).
5.1. Conclusions
In the current study, the genotypic and phenotypic features of infantile NS were displayed. NPHS1 mutations cause severe and early disease type but with better prognosis. Additionally,E117K polymorphism of NPHS1 showed a similar course as other NPHS1 and NPHS2 mutations, with the only difference being that E117K polymorphism manifested relatively earlier onset. Also, among NPHS1 mutations, E117K had been reported as a polymorphism, but we showed our contrary findings and ask: Is it still a polymorphism?
