
Fulltext
Holoprosencephaly (HPE) is a malformation that arises during the first 4 weeks of embryonic development (blastogenesis)[1] caused by a failure or incomplete division of the prosencephalon into cerebral hemispheres. This defect is frequently associated with other facial anomalies such as anophthalmia, cyclopia, proboscis, midface clefting, hypertelorism, single maxillary central incisor, and absence of olfactory nerves or corpus callosum. It is a causally heterogeneous field defect caused by: 1) chromosome aberrations in particular trisomy 13[2], partial deletion of the long arm of the chromosome 7, triploidy (69, XXY)[3] and other recessive, dominant, or X-linked genes[2] and multifactorial causes. Klinefelter syndrome is the most common sex chromosome abnormality in men and boys, with a reported prevalence of 0.1% to 0.2% in the general population and of up to 3.1% in the infertile male population[4]. Since the first report in 1942, Klinefelter syndrome has been characterized by small, firm testes and varying symptoms of androgen deficiency including gynecomastia, hypogonadism, infertility[5] and immaturity of external genitalia. Although additional X chromosomes are pre¬dominantly inactivated, the entire chromosome region is not inactivated, and inactivated region of the additional X chromosome is likely to be responsible for the clinical features[6].
A male neonate was born at full term (38 weeks of gestation). His mother was nulipara. His Apgar scores were 5 and 7 at 1 and 5 min respectively. He was the first child of healthy and non consanguineous parents. The family history was negative for abortion and genetic abnormalities. The pregnancy was uneventful. The antenatal ultrasonography revealed microcephaly and cleft deformity.
Assessment after birth showed body weight of 3200 g (25-50percentile), head circumference of 29.5 cm (<10 percentile) and body length of 50 cm (25-50percentile). His father and mother were both healthy, aged 25 and 30, respectively.
On physical examination, phenotypic abnormalities including bilateral cleft lip/palate and low-set ears, hypotelorism, exophthalmous, one sided nostril without columella, absence of premaxilla, underdeveloped septum and vomer (arinia) with microcephaly were observed (Fig. 1). The other physical and laboratory findings were normal in the cardiac, ventricular, and genitourinary systems. In neurologic evaluation he has generalized epilepsy that is controlled with 15 mg phenobarbital bid. The genitalia were normal without ambiguity, and both testes were palpable outside the inguinal rings. The karyotype of peripheral blood lymphocytes was 47,XXY/46,XY. The head CT scan revealed colpocephaly and overriding parietooccioital suturs (Fig. 2).
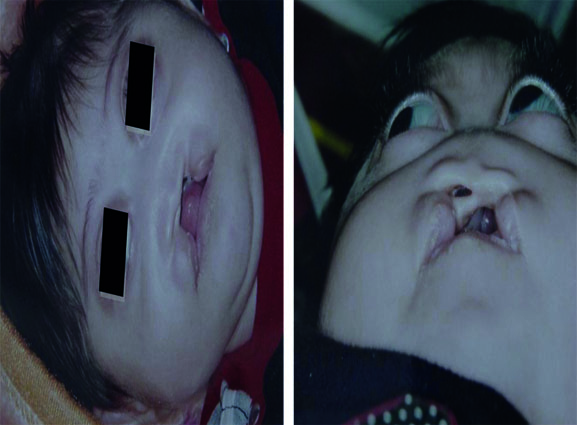
Fig.1: Holoprosencephalic patient photography from anterior (left), note to wide lip and alveolar cleft; and from below (right), note to midface deformity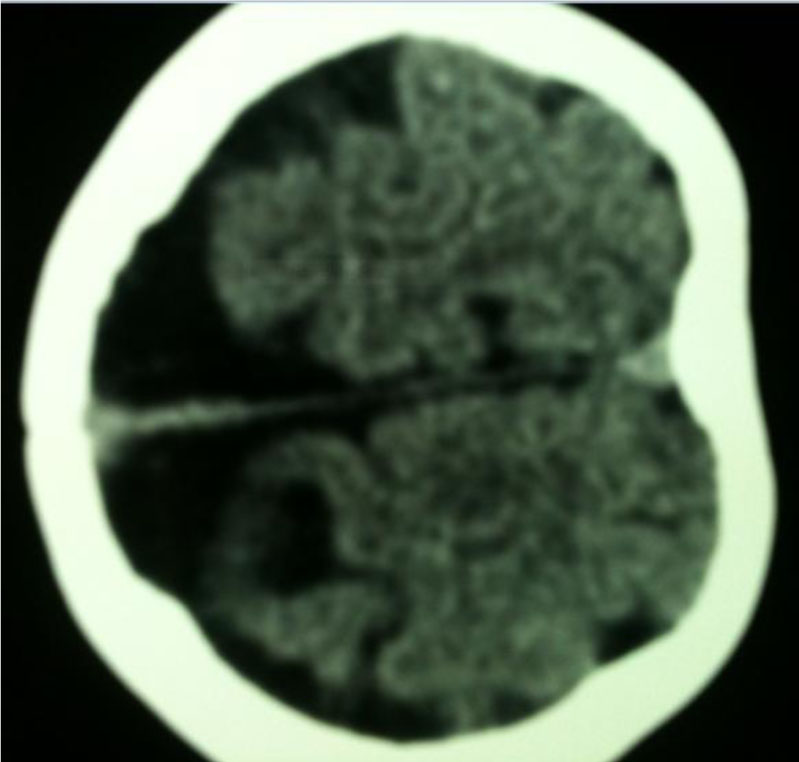
Fig. 2: Brain CT scan from patient with holo-prosencephaly and Klinefelter syndrome
Klinefelter syndrome may remain largely undiagnosed, unless there are typical clinical features and cytogenetic abnormalities. Most 47,XXY neonates appear normal at birth, and boys have variable phenotypic features without obvious facial dysmorphology[5]. There are three major variants of Klinefelter syndrome including 48,XXXY; 48,XXYY; and 49,XXXXYl[7].
Phenotypic abnormalities which may be observed in variant Klinefelter syndrome include: microcephaly with short stature, hypertelorism, flat nasal bridge, fifth finger clinodactyly, bifid uvula, heart defect, radioulnar synostosis and genu valgum[7]. To the best of our knowledge, cleft palate has been described in only one case with the 49XXXXY/46XY karyotype and one case with 48XXXY/46XY karyotype[8]. Therefore cleft palate might be a rare clinical presentation of the variant Klinefelter syndrome. Cleft palate is a common birth defect and is considered to be a multifactorial condition in which genetic predispositions interplay with environmental factors[9].
We describe a case of variant Klinefelter syndrome with 48XXXY/46XY mosaicism and cleft palate. There were 2 cases of cleft palate with varriant types of Klinefelter syndrome reported in the past. Our case entered in cleft workup after initial managements and genetic studies. We repaired his cleft lip and cleft palate in 2.5 and 12 months respectively (Fig. 3). One sided nostril and short clumella were reconstructed in combination with revision surgery. The patient ws hospitalized
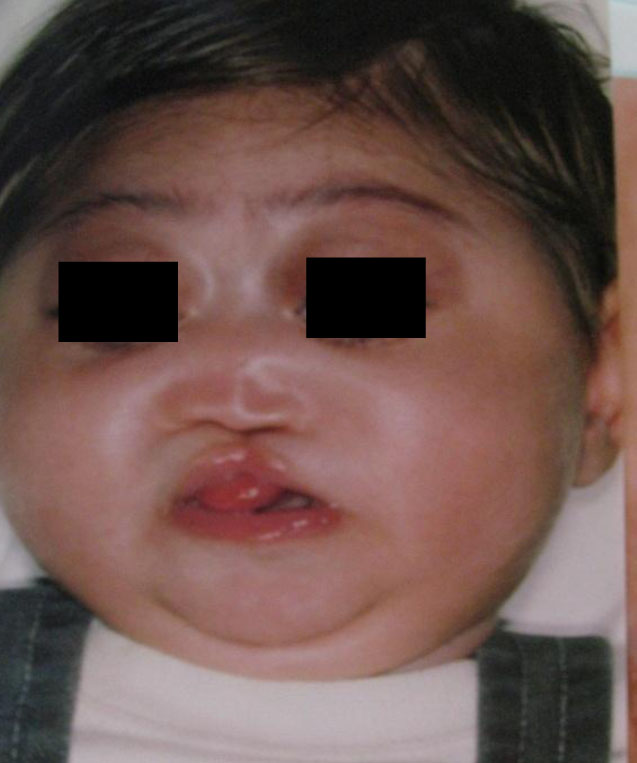
Fig. 3: Holoprosencephalic patient after lip and nose deformity reconstruction
several times because of respiratory infections. His dismorphic face with exophthalmia and midface growth retardation is planned to be repaired surgically in future.