



1. Background
Coronavirus disease 2019 (COVID-19) began in late 2019 in the Wuhan region of China. According to WHO reports, due to its high prevalence, it has affected a large number of people worldwide (1-3). To date, many people have died from this disease (4) . Evaluations of the virus SARS-CoV-2 have shown that this single-stranded RNA virus has multiple hosts. It is an enveloped, non-segmented, positive-sense single-stranded RNA virus genome, with a size ranging from 26 to 32 kbp. The virion contains a nucleocapsid composed of genomic RNA and a phosphorylated nucleocapsid (N) protein, which is buried inside phospholipid bilayers and covered by the spike glycoprotein trimer (S). The membrane (M) protein (a type III transmembrane glycoprotein) and the envelope (E) protein are located among the S proteins in the virus envelope, making it a recognized global health hazard (2).
To date, different drugs such as hydroxychloroquine (HCQ) (5), Azithromycin (6), and combinations of both have been used to treat COVID-19 infections. Several studies have demonstrated that chloroquine phosphate and chloroquine sulfate have the ability to inhibit COVID-19 in vitro (7-9). However, among the candidate treatments, only three main drugs, including Remdesivir, Oseltamivir, and HCQ, have been tested in large comparative studies (10-12). A few studies have shown that Lopinavir, Ritonavir, and Remdesivir have no clear influence on COVID-19 and are associated with many adverse effects (12-14).
Ivermectin is an inexpensive FDA-approved antiparasitic drug known for its broad-spectrum antiviral activity against a variety of viruses in vitro. It also inhibits the proliferation of COVID-19 cells in cell cultures (15, 16). Moreover, ivermectin has shown antiviral activity against several RNA viruses, including Zika, dengue, yellow fever, West Nile, Hendra, Newcastle, Venezuelan equine encephalitis, chikungunya, Semliki Forest, Sindbis, Avian influenza A, Porcine Reproductive and Respiratory Syndrome, and HIV-1. Additionally, studies have demonstrated its antiviral effects against DNA viruses such as equine herpes type 1, BK polyomavirus, pseudorabies, porcine circovirus 2, and bovine herpes virus 1 (17).
Ivermectin may act on the COVID-19 virus through mechanisms that reduce the viral load. These mechanisms include nuclear import inhibition (6) and interference with the attachment of the spike protein to the human cell membrane (18). Little viral replication occurs in the later phases of COVID-19 infection; the virus cannot be cultured, and only a minority of autopsies show viral cytopathic changes (19, 20).
Several studies have demonstrated the anti-inflammatory properties of ivermectin, including its ability to inhibit cytokine production after lipopolysaccharide exposure, down-regulate the transcription of NF-κB, and limit the production of both nitric oxide and prostaglandin E2 (21, 22). In vitro studies have shown that ivermectin decreases the activity of NF-κB, MAP kinases, JNK, and P38 (23). Additionally, studies have demonstrated the anti-inflammatory effects of the drug in animal models (24, 25).
2. Objectives
Therefore, to further investigate these mechanisms, a molecular dynamics (MD) simulation method was performed to better understand the mechanism of inflammation. In the present study, we aimed to evaluate the binding of ivermectin to NF-κB/MAPK10/MAPK14 proteins using molecular docking and MD simulation methods, which could provide useful information for the treatment of the disease.
3. Methods
3.1. Molecular Dynamics Simulation
The crystal structures of the nuclear factor NF-κB (entry code: 1LE5), Mitogen-activated protein kinase 14 (entry code: 4DLI), and Mitogen-activated protein kinase 10 (entry code: 4H39) were obtained from the PDB Bank (http://www.rcsb.org). The characteristics of ivermectin were provided by PubChem. To assess the allowed torsions for the ligand, characterize the search space coordinates, and add polar hydrogen atoms to the protein, the graphical AutoDock tool was used (26).
Afterwards, the docking process was performed with a grid size of 70 × 90 × 70 along the X, Y, and Z axes with 1 Å spacing for the NF-κB-ivermectin complex, 76 × 68 × 82 for the MAPK 14-ivermectin complex, and 70 × 64 × 80 for the MAPK10-ivermectin complex. The lowest binding energies of the NF-κB-ivermectin, MAPK 14-ivermectin, and MAPK 10-ivermectin complexes were determined using AutoDock Vina and were considered the primary structures for the MD simulation process (27).
The GROMOS 53a6 (GROMACS 5.1 package) was applied to carry out MD simulations of the complexes (28). The ProDrug program was used to provide the topological characteristics of ivermectin (29). In this study, the complexes were solvated using a transferable intermolecular potential with a 3-point (TIP3P) water model in a cubic box with a distance of 10 Å from the furthest atom of the protein (30). After solvation, Na+ and Cl− ions were added to neutralize the system. To solve the linearized Poisson–Boltzmann equation, at an ionic strength of 150 mM (NaCl), a solvent radius of 1.4 Å, and a relative permittivity of 80, a concentration of 150 mM NaCl was added to the systems (31, 32), and energy minimization was performed using the steepest descent method.
The equilibration process for each system was conducted with 1 ns MD simulation in the canonical (NVT) ensemble and 1 ns MD simulation in the isothermal–isobaric (NPT) ensemble using position restraints on the heavy atoms of the protein to allow for the equilibration of the solvent. The Nose–Hoover thermostat was used to maintain the temperature of the system at 300 K. To preserve the system pressure at a fixed 1 bar, the Parrinello–Rahman pressure coupling method was applied (33). The particle Mesh Ewald (PME) method with 1.0 nm short-range electrostatic and van der Waals cutoffs was used to measure the electrostatic interactions (34). Finally, a 100 ns MD simulation for each complex (NF-κB-ivermectin, MAPK 14-ivermectin, and MAPK 10-ivermectin) was performed with time steps of 2 fs on equilibrated systems.
3.2. Potential of Mean Force
Umbrella sampling (US) is a method used to provide the free energy profile, often referred to as the potential of mean force (PMF), along a specific reaction coordinate, such as protein-protein separation distance. Applying physical reaction coordinates can yield more structural insights (35). In the current study, the binding energies of the NF-κB-ivermectin, MAPK 14-ivermectin, and MAPK 10-ivermectin complexes were estimated from PMF using the US method.
First, the MD simulation was carried out to drive ivermectin far away from the protein, which was fixed during the simulation. Next, 50 configurations were created along the z-axis coordinate. The z coordinates of the center of mass (COM) interval between ivermectin and the proteins varied by 0.5 Å in each configuration with a force constant of 10 kcal/mol·Å. The equilibration process for each window was performed over a period of 10 ns, followed by a 10 ns production run for sampling (36, 37). Ultimately, the weighted histogram analysis method (WHAM) was used to create the PMF profile, executed by GROMACS using the ‘g_wham’ command (38).
To effectively analyze the MD process, the root mean square deviation (RMSD) and hydrogen bonds (H-bonds) were analyzed using GROMACS tools during the simulation. The final PDB file of the MD simulation was visualized using Pymol software. Additionally, to evaluate the H-bond and hydrophobic interactions of the aforementioned complexes, LigPlot software was used (39).
3.3. Molecular Mechanics Poisson-Boltzmann Surface Area Method
The molecular mechanics Poisson-Boltzmann surface area method (MMPBSA) has been extensively used to measure the affinities of molecular models, including ligand-protein and protein-protein interactions (40, 41). In the current study, the binding free energy between NF-κB (ligand-receptor) was measured during the equilibrium phase at an interval of 50 ps from 80 - 100 ns MD simulation using g-mmpbsa GROMACS software (42). The binding free energies (ΔG_bind) of each simulated spike and complex were calculated using the g_mmpbsa v5.12 package tool in the GROMACS platform. This post-simulation binding energy calculation is based on the molecular mechanics/Poisson Boltzmann surface area (MM/PBSA) approach. The Gibbs binding free energy (ΔG_bind) was calculated using the following equation:
Where ΔEMM is the vacuum potential energy, whichincludes the energy of both bonded (ΔEbonded) as well as non-bonded (ΔEnon-bonded = ΔEvdw + ΔEelec) interactions. TΔS indicates the entropic contribution to the vacuum-free energy.
Where ΔGsol is the free energy of solvation, and ΔGpolar and ΔGnonpolar are the electrostatic and non-electrostatic contributions to the solvation free energy, respectively.
4. Results
4.1. Molecular Docking and Molecular Dynamics Simulation Analysis
This study was designed to investigate the inhibitory effects of ivermectin on the NF-κB, MAPK10, and MAPK14 inflammatory pathways using docking and MD simulation studies. To determine the optimal binding manner of ivermectin to NF-κB, MAPK14, and MAPK10, a molecular docking process was carried out. The results demonstrated that ivermectin binds to a site located between chain E and chain A of NF-κB, with binding energies of -9.9, -8.2, and -9 kcal/mol, respectively. The best structure for the complex with the lowest binding energy was created using AutoDock Vina software, which was assumed to be an appropriate model for the MD simulation process.
To predict system equilibration during the simulation, RMSD was used as an appropriate parameter (43, 44). To ensure system equilibration, the RMSD profiles of the NF-κB–ivermectin complex were analyzed during the 100 ns simulation process. As shown in Figure 1, the RMSD profiles of the complexes were evident. The systems were equilibrated almost after 10 ns. The RMSD average of the NF-κB–ivermectin complex in the last 5 ns of the simulation was calculated as 0.35 nm. Protein residues are critical in generating a stable conformation for a protein-ligand complex, which can be assessed using the RMSF parameter. Higher RMSF levels indicate increased flexibility, suggesting an increased capacity to interact with the ligand molecule, while lower RMSF fluctuations indicate less flexibility and reduced interaction potential. Additionally, the backbone atoms of each amino acid residue of NF-κB and the NF-κB–ivermectin complex were analyzed (Figure 2). The protein compaction level was determined by the radius of gyration (Rg).
 value of NF-κB -ivermectin Cα alone and the Cα of NF-κB -ivermectin complex.")
The root mean square deviation (RMSD) value of NF-κB -ivermectin Cα alone and the Cα of NF-κB -ivermectin complex.
as a function of amino acid residues")
Root mean square deviation (RMSD)as a function of amino acid residues
The Rg is defined as the mass-weighted root-mean-square distance of a collection of atoms from their common COM. The trajectory analysis of the Rg indicated the evolution of the overall protein dimension throughout the dynamics. Since determining the relative distance of each atom from the protein's COM is complex, we used the Rg factor to analyze protein folding or unfolding. As shown in Figure 3, the Rg of NF-κB was greater than the Rg of the NF-κB–ivermectin complex at the end of the simulation time, indicating system equilibrium. The NF-κB–ligand structure showed four significant ascents at 53 ns. Within less than 19 ns, the Rg of complexation increased, related to hydrophobic interactions. The Rg increased from 1.58 to 1.65, making the system more open. The change in system state from folded to open indicates the system’s instability. After 50 ns, little change is observed, meaning the system is in equilibrium. For the ivermectin–ligand complex, the average Rg value is 1.6502 nm, with a significant ascent of about 3.2 ns (Figure 3). In contrast, the average Rg value for NF-κB is about 1.58 nm, with various decays in the Rg plot, as presented in Figure 4A.
In general, the MD results showed that NF-κB in the complex is more unstable than NF-κB in the free state, with evident fluctuations in the complex. Protein intermolecular H-bond formation plays an essential role in the stability of the system. The H-bond network is an exclusive solvent with the ability to monitor stability, structure, dynamics, and biomolecular functions (45). The average number of H-bonds between NF-κB and ivermectin during the last 5 ns of the simulation was 14 (Figure 5).
 as a function of simulation time")
Radius of gyration (Rg) as a function of simulation time

The average number of hydrogen bonds as a function of simulation time for NF-κB –ivermectin complex

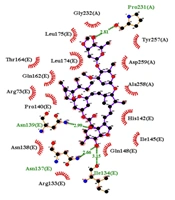
The interaction of NF-κB -ivermectin created by Lig-plot software. The 2D image of NF-κB -ivermectin complex.
4.2. Results of MM-PBSA Calculations
Furthermore, the complex's binding free energy was calculated using the MM-PBSA method (46). The binding free energy consists of contributions from polar solvation, van der Waals interactions, electrostatics, and SASA energy. Electrostatic, van der Waals, and SASA energies all contributed negatively to the overall binding free energy, indicating favorable interactions, while polar solvation energy contributed positively, indicating an unfavorable interaction. As demonstrated in Table 1, van der Waals energy was the primary contributor to the favorable binding interaction between NF-κB and ivermectin.
| Number | Total Binding Free Energy (kJ/mol) | Van der Walls Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Polar Salvation Energy (kJ/mol) | SASA Energy (kJ/mol) |
|---|---|---|---|---|---|
| Complex | ˜ -51 | ˜ -97 | ˜ -21 | ˜ 79 | ˜ -11 |
Molecular Mechanics Poisson-Boltzmann Surface Area Method Based Total Binding Free Energies Along with Its Constituent Energies for NF-κB with Ivermectin
Figures 4 and 6 demonstrate the 2D and 3D images of NF-κB in interaction with ivermectin after a 100 ns MD simulation. As illustrated, the 2D and 3D images of the NF-κB-ivermectin complex after 100 ns of MD simulation show significant interaction details. In the 2D image, ivermectin forms hydrophobic interactions with Arg73 (E), Arg133 (E), Asn138 (E), Pro140 (E), His142 (E), Ile145 (E), Gln148 (E), Gln162 (E), Thr164 (E), Leu174 (E), Leu175 (E), Gly232 (A), Tyr257(A), Ala258 (A), and Asp259 (A). Additionally, hydrogen interactions are observed with Ile134 (E), Asn137 (E), Asn139 (E), and Pro231 (A) residues (Figure 4). The results from both 2D and 3D structures demonstrate that ivermectin has more hydrogen and hydrophobic interactions with chain E of the protein than with chain A.

The interaction of NF-κB -ivermectin created by Pymol software. The 3D image of NF-κB -ivermectin complex.
4.3. Binding Energy Measurement
Umbrella sampling is a method widely used to measure free energy and evaluate the ligand-receptor separation process and binding free energy determination (47, 48). To identify the interaction between NF-κB and ivermectin, the PMF value was measured. The results of the PMF analysis are shown in Figure 7. The findings illustrate that ivermectin binds to NF-κB with a binding energy of -3.3 kcal/mol.

The profile of binding free energy of NF-κB dissociated from ivermectin.
5. Discussion
Computational determination of the binding modes of ligands with their targets by molecular docking is commonly employed in different drug designs (49). Coronaviruses are noteworthy because they possess various protected non-structural and structural proteins that have potential applications in drug design and discovery.
It has recently been reported that ivermectin may be a clinically useful anti-inflammatory agent for late-stage COVID-19 disease (23). In fact, ivermectin suppresses the activation of both NF-κB and stress-activated MAP kinases JNK and p38, making it a powerful strategy for the design and development of potential anti-inflammatory agents (21). Additionally, ivermectin is a potentially multifunctional drug that is absorbed through the intestine and then processed in the liver without any toxicity. This drug is not permeable to the blood-brain barrier and has shown stability against temperature changes (50).
Various medications have been tested, as previously indicated, but ivermectin has shown effectiveness in various clinical trials (51, 52). In-vitro and animal studies on ivermectin's antiviral activity suggest its efficacy in preventing and treating infections in the early stages. However, the concentrations examined in these in-vitro studies were more than 50-fold higher than the typical Cmax achieved with a standard single dosage of IVM at 200 µg/kg, raising concerns about the effective dose of IVM for treating COVID-19 in humans and its tolerability [Chaccour et al. (as cited by Niaee et al.)]. The findings of clinical trials and cross-sectional investigations conducted by this study team to determine the dose of ivermectin in mild COVID-19 patients revealed that a single dose is more effective than an interval dose compared to the control group (52, 53). Additionally, ivermectin, compared to two other drugs, HCQ and azithromycin—which have side effects such as myopathy, neuropathy, and retinal damage—is cheaper and more cost-effective (54).
Focusing on investigating the basic mechanisms governing ivermectin inhibition of pro-inflammatory cytokine production and clinical trials seems to play an important role in advancing previous findings. So far, several studies on the mechanisms of inhibition of Ivermectin–SARS-CoV-2 have been reported. One study showed that ivermectin has significant binding affinity with NSP3, NSP10, NSP15, and NSP16, which helps the virus evade the host immune system. Moreover, for further investigation, the MD simulation method was performed to better understand the mechanism of inflammation. For example, the results of the MD simulation study showed that the binding of ivermectin to Mpro, Spike, NSP3, NSP16, and ACE2 was fully stable (55).
Coronaviruses consist of four structural proteins called Spike, Envelope, Nucleocapsid, and Membrane proteins. SARS-CoV-2 enters human host cells through the binding of the RBD fragment of the spike protein to the angiotensin-converting enzyme 2 (ACE2) receptor (56, 57). This receptor is expressed in many tissues, such as the heart, kidney, and intestine, and these cells express genes related to viral replication, leading to the replication of the virus in the host lungs (58, 59). Recent studies showed that the ACE2 receptor is highly expressed in type 2 alveolar epithelial cells; in fact, SARS-CoV-2 uses these cells for invasion and proliferation (59).
Marik et al. (51) demonstrated in a simulation study that 25-hydroxyvitamin D is more effective in treating COVID-19 compared to lopinavir, showing the strongest interaction with the possible binding sites of the SARS-CoV-2 protein. Another study identified both hydrophobic and hydrophilic interactions in anchoring ivermectin inside the binding site of Nsp9 as well as the main binding track of the IMPα (60). Additionally, it was reported that ivermectin, by reducing compression and unfolding of two crucial proteins in SARS-CoV virus replication, such as 3CL protease and the HR2 domain, exhibited an inhibitory effect and could promote significant structural changes in these proteins (61).
Ci et al. (21) conducted a laboratory study indicating that ivermectin could inhibit LPS-induced pro-inflammatory cytokine production. These effects might be mediated by down-regulating the phosphorylation of ERK1/2, JNK, and p38 MAPK signaling and by inhibiting the activation of NF-κB pathways. They also showed that ivermectin treatment significantly inhibited p38 (MAPK-14) and JNK phosphorylation in a dose-dependent manner (21).
Extensive MD simulations have shown that the impressive destabilization of the RBD/ACE2 complex in the presence of ivermectin supports a direct inhibition of SARS-CoV-2 entry into the host cell. Moreover, inhibition of the active site of both 3CLpro and PLpro viral proteases could also contribute to inhibiting viral maturation after infection (62). A comparison of molecular docking of ivermectin and doxycycline showed that ivermectin has a greater binding affinity to virus proteins such as Mpro, spike, PLpro, RdRp, nucleocapsid, NSP3, NSP9, NSP10, NSP15, NSP16, and the host ACE2 receptor (55).
Based on previous findings, this study aimed to evaluate the binding of ivermectin to the NF-κB/MAPK10/MAPK14 proteins using molecular docking and MD simulation methods, which could provide useful information about the treatment of the disease. Due to the anti-inflammatory role of ivermectin, this study aims to better understand its binding mechanisms to other inflammatory proteins, suggesting it as an appropriate candidate to treat COVID-19. However, further investigations are required.
5.1. Conclusions
Ivermectin is a broad-spectrum antiparasitic drug with proven anti-inflammatory effects in vitro, though its targeting mechanism remains unclear. In the present study, the affinity of ivermectin was evaluated using docking, MD simulation, MMPBSA, and PMF methods. The results demonstrated that ivermectin binds to NF-κB with high affinity, suggesting that this protein can be an appropriate drug target to decrease inflammation, thereby confirming the anti-inflammatory activity of ivermectin. Thus, it could be suggested that ivermectin may be a good candidate for treating SARS-CoV-2 infections.
