



1. Background
Coronaviruses (CoVs) are RNA-viruses that belong to the Coronaviridae family which invade the human respiratory system and cause a variety of respiratory infections (1). Other members of this family include severe acute respiratory syndrome (SARS)-CoV and the Middle East respiratory syndrome (MERS)-CoV, which also can cause major respiratory problems (2). The first case of SARS-CoV-2 was reported on December 12, 2019, in Wuhan, Hubei Province, China. With a death rate of 3.4%, it has claimed about 24 257 989 lives worldwide, until August 28, 2020 (3). The World Health Organization (WHO) is trying to control the pandemic and reduce the mortality rate (4, 5).
Currently, there is no treatment for COVID-19; however, immediate measures are needed. Previous studies were mostly focused on developing novel therapeutics mediators, including antivirals drugs and vaccines (6). Interferon (IFN)-α and ribavirin are among the most important therapeutic mediators that are under investigation (7). Based on the currently available data, the following therapeutic options are proved to be effective against COVID-19 infection: Ritonavir (8), lopinavir (8, 9), either alone or in blend with remdesivir (10, 11), oseltamivir (9), chloroquine (12, 13). Among these drugs, ritonavir, remdesivir, and chloroquine are reported to be more effective in reducing the severity of the symptoms (12); however, further evidence are needed (14).
The SARS-CoV-2 is an enveloped positive-stranded RNA virus with ~30,000 nt RNA genome (15, 16). The coronavirus replicase gene expression involves two overlapping polyproteins, named pp1a and pp1ab (17, 18). These polyproteins are divided into mature non-structural proteins, including the main protease (Mpro) and a papain-like protease, in which all of them perform important functions in viral replication and transcription processes (6, 18).
To control viral gene expression and replication, a highly complex network of proteolytic cascades on the polyproteins is required (19). The main protease of the coronavirus mediates this maturation process (20). The N-terminal of Mpro has an important role in the proteolytic activity, and its C-terminal is necessary for dimerization action (21). It’s also suggested as a therapeutic option for SARS-CoV-2 (6, 20). Generally, some biological active peptides are engaged in the immune system of mammals (22-24) and through eukaryotic cells create protection against pathogens such as viruses, bacteria, and fungi (25). Research on the therapeutic activities of natural peptides dates back to the 1970s (22). Such peptides can be originated from natural sources, such as animals, plants, or microorganisms (26). The structure of peptides determines their therapeutic mechanisms. Peptides can be formulated to mimic the ligands or interact with the conserved domain in the protein surface by various software. The peptide sequence can be manipulated to achieve the highest therapeutic efficiency (27). The efficacy, safety, selectivity, and predictability of peptide drugs should be analyzed by in silico methods and to find the appropriate peptides inter to the in vivo demonstration steps (22).
The Pacific oyster, Crassostrea gigas, is a mollusk that naturally presents in marine environments (28). The hemocytes of this oyster, immunocompetent cells, creates an essential object in innate antimicrobial immunity responses. Also, they can produce antimicrobial peptides (AMPs) and release factors such as lectins and reactive oxygen species (ROS), which there are reports on their in vitro and in Molecular Docking method, deals with the strength, properties and specificity of binding a small molecule as a ligand to a larger molecule that acts as a receptor. These computational methods provide information on the binding activity and affinity of ligands and receptors (29).
2. Objectives
Several studies have investigated the effectiveness of different molecules against the new virus main protease; in this line, the current study intended to screen the inhibitory effect of Pacific oyster, Crassostrea gigas two antimicrobial polypeptides on the SARS-CoV-2 main protease, using computational docking techniques, because these polypeptides have an inhibitory effect on HIV-1 protease in vitro situations (30, 31).
3. Methods
3.1. Receptor and Ligand Preparation
The crystallographic structure of the target receptor, main protease (Mpro) protein of SARS-CoV2 in complex with an inhibitor N3, was retrieved from RCSB Protein Data Bank (PDB ID: 6LU7) (32) (https://www.rcsb.org/). In the docking performance step, the PDB file was imputed to Molegro Virtual Docker (MVD 6 edition, a CLC Bio Company, Denmark) software (33) (Figure 1). The first optimization was performed by adding hydrogen atoms, because most of the macromolecular structure data do not contain hydrogen atoms in their corresponding PDB files. The water molecules were removed to make computations easier and to better clear the binding pocket of possible water molecules that would distort the pose search. Remember is a molecule that can create multiple favorable contacts to the protein, water molecules might confound this procedure (Figure 2). This procedure can remarkably increase the calculations and to evade any expected deformity (34). In the PDF file downloaded from the PDB database (ID: 6Lu7), the main protease (Mpro) of SARS-Cov2 binds to an inhibitor called N3. and should be removed when preparing the protein in the pre-analytical process. Therefore we removed this internal ligand from protein. Next, water molecules were removed, and the protein was prepared through the MVD molecule preparation step. The discovery of functional cavities was then applied to find the excellent docking constraints on the protein structure. Four cavities were found, and the fifth had an extra resemblance to the fourth cavity. We selected two Pacific oyster, Crassostrea gigas antimicrobial polypeptides, including HIV-1PIP-1 (HIV-1 polymerase inhibitory polypeptide 1) and HIV-1PIP-2, with amino acids sequences Leu-Leu-Glu-Tyr-Ser-Leu and Leu-Leu-Glu-Tyr-Ser-Ileu, respectively, were selected from the antivirus lists and according to the literature (35-38). These Pacific oyster polypeptides have been documented to have inhibitory effects on HIV-1 polymerase in vitro. Besides, their antiviral effects have been approved in the previous publications (39-42). Similar to various chemicals used to inhibit SARS-CoV-based on antiviral polymerase compounds, we selected these compounds to dock and compare the results with reference compounds such as remdesivir (43-46). The two dimensional (2D) structure, molecular formula, molecular weight, and source of these ligands were obtained from the National Center for Biotechnology Information as well as Explore chemistry databank system PubChem (https://pubchem.ncbi.nlm.nih.gov/) (Table 1).
.")
A, Visualization using Molegro Virtual Docker hydrophobicity surface and B, secondary structure of the protein (6lu7).
 which was used to specify the docking constraints, dimensions, and coordinates (B). The coordinates properties are: X = -10.76, Y = 12.64 and Z = 68.81.")
The target cavity (A) which was used to specify the docking constraints, dimensions, and coordinates (B). The coordinates properties are: X = -10.76, Y = 12.64 and Z = 68.81.
| Ligand Name | MolDock Score | Rerank Score | Total Interaction Energy (Between Energy) | Torsions | HBond |
|---|---|---|---|---|---|
| Remdesivir | -161.419 | -98.9254 | -172.705 | 14 | -6.03673 |
| HIV-1PIP-1: Leu-Leu-Glu-Tyr-Ser-Leu | -158.545 | -107.821 | 171.915 | 23 | -6.44185 |
| HIV-1PIP-2: Leu-Leu-Glu-Tyr-Ser-Ile | -143.864 | -106.631 | -171.402 | 23 | -3.69101 |
The MVD docking software was used. The main protease structure, along with all selected ligands, were picked out for docking procedures. The selective and best binding site constraints were fixed to comprise the largest detected cavity on the main protease protein structure and, then, its sizes were reduced to achieve the desired simulation treating time and increase the accuracy. Then the best docking cavities of the receptor were found, and an excellent docking grid was prepared, with a radius of 10 (Å) and coordinates of X: -10.76, Y: 12.64 and Z: 68.81. In each ligand, five poses were docked with 10 runs. In the present study, the biggest cavity was selected for the docking process, because in the previous studies this cavity is considered as a major candidate for the docking process and as a major region that ingredients can inhibit this protein for coding a major functional structure.
3.2. Docking Process and Analysis
The docking process of the two selected polypeptide was carried out in comparison to remdesivir, as a comparative standard for molecular docking and MD simulation analysis. Using computational docking techniques, we selected two antimicrobial polypeptides on the SARS-CoV-2 main protease, based on their proven in vitro inhibitory effect on HIV-1 protease.
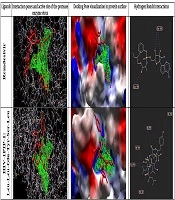
After docking, the results were imported into the Molegro Molecular Viewer (MMV 2.5 edition, a CLC Bio Company, Denmark) software to visualize the 2D structure of each ligand (47). The best pose of each ligand was selected based on scores of MolDock Score, Rerank Score, total interaction energy (between energy), and HBond items. For the best poses, the 2D diagrams of receptor-ligand interaction, interaction poses, the active site of the virus protease enzyme, protein surface, and Hydrogen Bonds were visualized using Molegro Molecular Viewer. Based on MVD software report, the RMSD score was less than 1.
4. Results
The cavity divination by mathematical tools revealed the presence of four cavities on the Mpro surface with an area of about 10.24 - 126.98 Å. The biggest and wider cavity was selected for docking, as it primarily contained the binding ligand 02J-ALA-VAL-LEU-PJE-010 in the used PDB file. The two selected peptide ligands (Figure 3) were appraised for binding likelihood in the same docking session.

Docking pose visualization
The results of the docking process were evaluated based on several bioinformatics docking scores, including MolDock score, Rerank score, Total interaction energy (between energy), and HBond (Table 1). The best polypeptide ligand was selected based on the MolDock score item ranking. Based on the results, the best ligand was HIV-1PIP-1 with amino acid sequences of Leu-Leu-Glu-Tyr-Ser-Leu, which showed a MolDock score closer to the control drug remdesivir (Table 1). Also, the Rerank score showed that the selected ligand has higher binding power and affinity to its receptor compared to the standard compounds of remdesivir, and this bonding strength can be increased by minor molecular modifications. It worth noting that HIV-1PIP-1 (Leu-Leu-Glu-Tyr-Ser-Leu) ligand was similar to the remdesivir concerning the total interaction energy and hydrogen bound or HBond scores (Figure 3). The graphical visualization showed that hydrogen bonds interact with a protein molecule. In all selected items for docking and standard form, the maximum hydrogen bond interaction between ligand and proteins was related to the HIV-1PIP-1 (Leu-Leu-Glu-Tyr-Ser-Leu) ligand, and All the amino acids that interacted with the ligand in the protein structure were visible. (Asn142, Ser144, Cys145, Glu166, Gln192).
5. Discussion
Developing potent and safe drug candidates against viruses like COVID-19, which has turned into a global crisis, is the main goal for drug development programs. Generally, some biological active peptides originated from natural sources, such as mammal tissues and animal venoms, or those with artificial sources have an important role in the immune system of mammals (22-24). These peptides can be potential candidates for protecting eukaryotic cells against a wide spectrum of pathogens, including viruses (25).
The Pacific oyster produces defensive peptides against environmental pathogens (28). In several studies, the antimicrobial role of these peptides has been investigated in vitro and in silico (48-50). In the present study, the binding affinity of the two selected defense peptides of Pacific oyster to coronavirus main protease was investigated.
As mentioned before, in the present study, we used the MolDock score for interpreting COVID-19 protease interaction with the peptides. Because MolDock is a fast algorithm, virtual screening was performed by using this scoring function. The MolDock is based on a new heuristic search algorithm that causes differential developments with a cavity prediction algorithm (33). In total, four cavities were found in the three-dimensional structure of the protease and the largest one was linked to its ligand in the PDB file. Therefore, it was selected for docking with the designed polypeptide ligands. The docking scoring function of the MolDock is an extension of the piecewise linear potential (PLP), including new hydrogen bonding and electrostatic terms (33, 51-53). To further improve the accuracy of docking, the re-ranking scoring function was used (33, 51, 53). The docking accuracy of the MolDock was evaluated by docking flexible ligands to 77 protein targets. MolDock could identify the correct binding mode in 87% of the complexes (54). Therefore, the findings of this method are highly accurate and interpretable (33, 51, 53). The data obtained through screening suggested that HIV-1PIP-1 (Leu-Leu-Glu-Tyr-Ser-Leu) polypeptide ligand can bind to Mpro with an affinity of -158.545 based on MolDock score and total interaction energy of 171.915. The remdesivir MolDock score was -161.419, and its score was very close to the above-mentioned polypeptide, which is an appropriate score for controlling the entropy. Therefore, in major index in industrial comparing in docking process, but as an interesting result, we showed that hydrogen bond of remdesivir was equal to -6.03673 and in HIV-1PIP-1 (Leu-Leu-Glu-Tyr-Ser-Leu) polypeptide was -6.44185; this is an important item in docking properties which can help us to focus on this polypeptide when using inhibitory roles against Mpro of SARS-CoV2. The hydrogen bonds between remdesivir and Mpro are in Glu166 and Cys145 amino acids, but in HIV-1PIP-1 (Leu-Leu-Glu-Tyr-Ser-Leu) polypeptide and Mpro are in Glu166, Ser144, Cys15, and Asn 142. These scores indicate that our designed potential inhibitor can efficiently bind to the structure of the Mpro (Figure 3), which indicates the best pose of the chemical inhibitor in contact with the Mpro structure and interactions between top poses and Mpro. In this figure, contact residues are determined. Compared to the remdesivir, which its effectiveness in viral protease inhibition function is well-documented, the HIV-1PIP-1 (Leu-Leu-Glu-Tyr-Ser-Leu) ligand has high binding strength to main COVID19 protease Mpro. This binding power was even higher than the control drug in the Rerank score. Also, the total interaction energy of this ligand was close to the control drug. Most importantly, the estimated numbers associated with the hydrogen bonding strength of the peptide designed were greater than those for the control drug, which indicates the probable superiority and effectiveness of this ligand in the protease inhibition. While the rerank-score in MVD provides an estimate of the strength of the interaction; however, it is not calibrated to the chemical units and it does not consider complex contributions (e.g. entropy) (51). Also, the rerank score of remdesivir was low, which indicates that unrolled gestures may a good choice for the docking process and targeting preparing goals. The total interaction energy of our compound are very similar, and this issue revealed that all of pose and ligand interaction energy are suitable to good interaction in the docking process. Ligand torsion number is related to the flexibility of ligand and generally incorporated as a crucial variable in the thermodynamic function of binding free energy, according to this issue, the Leu-Leu-Glu-Tyr-Ser-Leu ligand has higher torsion than remdesivir. Therefore, it can be argued that this substance is more appropriate for the docking process. Therefore, the HIV-1PIP-1 (Leu-Leu-Glu-Tyr-Ser-Leu) ligand can be evaluated as a drug precursor. In general, the HIV-1PIP-1 (Leu-Leu-Glu-Tyr-Ser-Leu) ligand is highly similar to standard drugs and can be considered as an appropriate and effective synthetic option. However, further studies are needed.
5.1. Conclusions
This study demonstrated that the HIV-1PIP-1 (Leu-Leu-Glu-Tyr-Ser-Leu) polypeptide isolated from Pacific oyster, Crassostrea gigas can be a potential inhibitory compound versa Mpro of SARS-CoV2, and it can be used for subsequent laboratory studies.
