



1. Background
Congenital central hypothyroidism (CCH) is a rare condition characterized by a hypothyroid state, which is associated with permanent thyroid-stimulating hormone (TSH) deficiency of a normal thyroid gland (1, 2). It causes by structural or functional disorders affecting the hypothalamic TRH neurons or pituitary thyrotrope cells and ultimately causing defective stimulation of TSH secretion. CCH occurs in approximately 1 in 20 000 to 1 in 65 000 live births worldwide (3). In contrast to primary hypothyroidism, the diagnosis of CCH can be more difficult due to not being covered by most of the neonatal screening programs that mainly use the detection of serum TSH levels. Thus, most CCH patients are diagnosed late when the children reach three years of age, which in some cases causes severe neurodevelopmental retardation and growth failure (4, 5). Apart from syndromic CCH causes, isolated CCH can be caused by mutations of the TSH-beta subunit (TSHβ), the immunoglobulin superfamily member 1 (IGSF1), or less frequently, thyrotropin-releasing hormone (TRH) receptor genes (2).
TSH is a member of the glycoprotein hormone family, which is a heterodimeric molecule consisting of a common alpha subunit and a specific beta subunit that confer proper biologic effect of hormones. The human TSHβ subunit gene is located on the proximal short arm of chromosome 1 (1p13) and contains 3 exons. Exon 1 is 5' untranslated, and exons 2 and 3 encode a 138-amino acid peptide. The twenty N-terminal amino acids encode a signal peptide that is cleaved to yield a mature protein of 118-amino acid detectable in serum (6). So far, more than 90 allelic missense variants in the TSHβ gene have been reported, whereas over ten mutations have been identified in CCH (Figure 1), including missense (G29R, C85R, C88Y, C105R, C108Y), nonsense (p.E32*, p.Q49*, and p.Q69*) and frameshift mutations (p.C125Vfs*10 and p.F77Sfs*6) (7-13), as well as two splice-site mutations (c.162G>A, c.162+5 G>A) (14). Also recently, a homozygous TSHβ deletion was reported (T313del) (15). These missense variants are located in different regions of the coding exons of the gene and disrupt important disulfide bridges essential for heterodimeric integrity or disrupt the conserved CAGYC region.
.")
2. Objectives
In this study, using a polymerase chain reaction (PCR) based direct sequencing strategy, we have described three different missense mutations in the TSHβ -subunit gene in 5 (out of 7) nonrelated Iranian patients affected by congenital TSH-deficient hypothyroidism.
3. Methods
3.1. Patients
Blood samples of seven children with CCH symptoms who had unrelated families were collected. The study population contained four males and three females with an age range of 3.0 - 6.5 years (mean age = 4.42). They were invited to the metabolism and endocrinology clinic affiliated with the Shahid Sadoughi Hospital in the city of Yazd (Iran) for molecular analysis of the TSHβ gene. The baseline clinical characteristics of the unrelated patients at the time of diagnosis of hypothyroidism are shown in Table 1.
| Patient IDa | Female/Male | Age (years) | Weight (kg) | Family History | TSH (mU/L)b | Free T4 (ng/dL)c | Total T3d (ng/dL) | Observed Mutation |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 6.0 | 14.3 | No | 0.6 | 0.23 | 25 | c.32T>A (p. F11Y) |
| 2 | F | 3.8 | 9.5 | No | 0.5 | 0.35 | 15 | c.40 A>G (p.T14A) |
| 3 | M | 3.0 | 9.1 | No | 0.7 | 0.45 | 10 | c.40 A>G (p.T14A) |
| 4 | M | 4.0 | 10.6 | No | 0.7 | 0.65 | 20 | c.40 A>G (p.T14A) |
| 5 | F | 3.5 | 8.2 | No | 0.5 | 0.5 | 25 | c. 316G>C (p.G106R) |
| 6 | M | 6.5 | 16.3 | No | 0.8 | 0.55 | 10 | - |
| 7 | F | 5.0 | 15.9 | No | 0.7 | 0.15 | 10 | - |
Clinical Phenotypes, Biochemical Parameters, and Observed Mutations in Seven Children with CCH.
The biochemical diagnosis of CCH is usually suspected by low serum free thyroxine (FT4) accompanied by a normal or low TSH concentration. In the present study, the diagnosis of CCH was based on the serum TSH level (TSH of less than 0.1 mU/L, with a normal limit of 0.5–5.0), free thyroxine (FT4: 0.4 ng/dL, with a normal limit of 0.7–2.5), and total levels of triiodothyronine (T3: less than 30 ng/dL, with a normal range of 70–170). Daily doses of 30 – 70 μg of L-thyroxine were administered to these children with CCH to raise the serum-free T4 above the normal range.
Thyroid hypoplasia, agenesis, and ectopic thyroid abnormalities were excluded in all cases via ultrasound. Furthermore, we assumed that children with CCH have higher levels of TSH than their counterparts with isolated CCH. Also, FT4 levels were significantly lower in children with CCH compared to those with isolated congenital hypothyroidism. Other pituitary functions were normal and MRI hypothalamic-pituitary area was performed, and the results were normal.
Therefore, the participants diagnosis of CCH was confirmed using laboratory results. Informed written consent was obtained from all participants. Besides, the study was approved by the Ethical Review Board of the Yazd University, and all procedures in the present study were following the 1964 Helsinki declaration and ethical standards. To evaluate whether the patients have heart congenital defects or even other known genetic syndromes or not, we reviewed their medical records. Also, 60 healthy subjects from the same geographic area were used as normal controls (35 males and 25 females, aged 5-10 years, Mean ± SD = 6.62 ± 1.7). Genomic DNA was extracted from peripheral blood leukocytes of the affected children and healthy controls using standard techniques (Qiagen Blood DNA kit, Hilden, Germany). The entire coding region of the TSH-β gene (exons 2 and 3) and the surrounding intronic sequences were amplified by the PCR and, then, products were purified and directly sequenced.
3.2. Amplification and Sequencing of TSHβ-Subunit Gene
DNA fragments containing the entire coding region of the TSHβ subunit gene (The human TSHβ subunit gene is organized into 3 exons: exon 1 is untranslated, and exons 2 and 3 encode a 138‐amino acid peptide) were amplified by the PCR using the specific primers 5'-TTCTTGGTTTCTTTGCCCTTTC-3' and 5'-CCGTGTCATACAATATCCAGC-3' for exon 2 and 5'-CCAGGATATCAATGGCAAAC-3' and 5'-TTCAGGCAAGCACATTTAACC-3' for exon 3. Particular primers were designed by the Primer1 online software (http://primer1.soton.ac.uk/primer1.html), based on human TSHβ sequence reported in the National Center for Biotechnology Information (NCBI). The PCR was performed in a total volume of 25 µL of the reaction solution containing 50 ng genomic DNA, 5 pmol of each primer, 0.5 mmol/L of each dNTPs, and 1.7 mmol/L MgCl2 in 1×PCR buffer and 0.4 U Taq DNA polymerase (Qiagen Company, Germany). Two independent PCR reactions were performed for coding regions of the second and third exons on a thermocycler (Eppendorf, Hamburg, Germany) as following: The PCR reaction of coding exon 2 initiated with 4 minutes at 94ºC, followed by 30 cycles of 30 seconds 94ºC, 30 seconds at 60ºC, 50 seconds at 72ºC, and finished with a 5 minutes final extension step at 72ºC. The PCR reaction of coding exon 3 was performed with an initial 94°C denaturation for 5 minutes, followed by 30 cycles of 94ºC for 30 seconds, 58°C for 30 seconds, and 72ºC for 35 seconds, terminating in a final extension at 72°C for 10 minutes. The quality of PCR products was analyzed using 1.5% agarose gel electrophoresis. To identify mutations, DNA direct sequencing of all subjects was performed by a commercial agency (Macrogene Seoul, South Korea).
3.3. In Silico Analysis
The online multiple sequence alignment software (MEGA4; available at https://www.megasoftware.net/mega4/) was used to find the degree of the homology of the sequence of TSHβ- subunit gene (gene ID: NG_015891.1, and transcript ID: NM_000549.5) that has been found in this study with sequences of the other species. Also, sequence alignment was performed using the Standard Protein Blast (blastp) program available at the National Center for Biotechnology Information (NCBI) website (http://www.ncbi.nlm.nih.gov/Blastp). Polymorphism phenotyping version 2.0.6 (PolyPhen-2) (http://genetics.bwh.harvard.edu/pph2/) and Sorting Intolerant from Tolerant (SIFT) (http://sift.jcvi.org/) were used to predict the functional consequences and pathogenicity effects of missense mutations using default settings. Also, the degree of protein hydrophobicity was predicted using a plot created by the Expert Protein Analysis System (ExPASy) (http://web.expasy. org/protocol). Eventually, as the structure of human TSHB protein has not yet been predicted, the homology-based structural prediction service SWISS-Model (https://swissmodel.expasy.org/) was used to generate the structure by submitting the human TP2 sequence (UniProt: P01222). Then, the effect of altered residue in the TSHB protein structure was evaluated by PyMol software, which is a visualization tool that can create high-quality 3D images of small molecules and biological macromolecules, such as proteins.
3.4. Statistical Analysis
SPSS version 20 (SPSS Inc., Chicago, Il, USA) was used to analyze the data. Statistical significance was considered when P value < 0.05.
4. Results
In this study, seven CCH pediatric patients (four males and three females) with a mean age of 4.42 ± 1.2 were recruited and clinically evaluated against a cohort of 60 ethnically matched unrelated healthy cases who were used as controls. The youngest and oldest participants were 3 and 6.5 years old. Amplification of the complete coding exons of the TSHβ gene was performed using the PCR. Direct DNA sequencing revealed three nucleotide changes: two novels heterozygous (c.32T>A and c.316G>C) and one homozygous (c.40A>G) missense mutations in the coding sequence of exons 2 and 3 in five unrelated patients (Figure 2).
 in 3 pediatric patients; C, heterozygosity for the other mutant allele (c.32T>A) in one patient; and D, heterozygosity for the third mutant allele (c.316G>C) in one patient.")
Electropherograms of the analyzed TSH-β sequences. A, Normal allele in a Control case; B, homozygosity for the mutant allele (c.40A>G) in 3 pediatric patients; C, heterozygosity for the other mutant allele (c.32T>A) in one patient; and D, heterozygosity for the third mutant allele (c.316G>C) in one patient.
One of the new heterozygous mutations (c.32T>A) and a homozygous mutation (c.40A>G) were found in exon 2 of the TSHß gene, causing amino acid substitutions in the signal peptide of the TSHß-subunit. The c.32T>A missense mutation at codon 11 of exon 2 converts the amino acid phenylalanine to tyrosine (F11Y) in the hydrophobic core of the signal peptide, and c.40A>G mutation at codon 14 of exon 2 changes the amino acid sequence of threonine to alanine (T14A) in the polar COOH terminal region of the signal peptide. For the novel F11Y missense mutation, the patient was heterozygous (a six-year-old boy), and for variation T14A, three patients were homozygous (two boys who were three and four years old and a 3.8-year-old girl). The c.40A>G nucleotide change (T14A, rs10776792) appears to be the most common genetic variation associated with congenital TSH deficiency (16). Based on the results of the NHLBI Exome sequencing project, this variation is a benign single nucleotide variant, and 6189 individuals (out of 6500 exomes sequenced) had GG genotype at this particular position. In the present study, this variation was not observed in any of the healthy and control subjects. However, some of the unaffected and clinically normal parents were heterozygous for T14A mutation (Figure 3). These pediatric patients did not have any siblings.

Pedigree aligned with sequencing results in parents of an affected boy with homozygous mutant allele: c.40A>G, p. T14A. Both parents are heterozygote for this mutation.
Also, the novel missense mutation (c.316G>C) in exon 3 is found in one pediatric patient with congenital central hypothyroidism. This mutation at codon 106 of exon 3 resulted in a change from glycine to arginine. This patient was a 3.5-year-old girl diagnosed based on the TSH, free fT4, and total T3 levels, which are described in Table 1. Since neurodevelopmental delay is common in patients with TSHβ mutations, in the present study, neurodevelopmental retardation and growth hormone deficiency were most obvious in this affected girl, thus special schooling services should be provided to her.
The molecular assessment of 60 individuals from the general population using the PCR-direct sequencing did not reveal the presence of any of the mutant alleles, indicating that these mutations occur in less than 1.6% of the population; P < 0.05. According to the ExPASy prediction results, these three alterations in the amino acid sequence might impair the proper function of the TSHβ protein, because they change the hydropathy determinants of the protein. Also, several functional prediction tools (structure and sequence homology-based approaches) predicted that these mutations may be harmful and affect protein function with a highly deleterious tolerance score < 0.01 (Table 2).
| Missense Mutation | c.32T>A, p. F11Y | c.40A>G, p. T14A | c.316G>C, p. G106R |
|---|---|---|---|
| Mfold Server | |||
| ∆G = -208.78 (native) | -207.68 | -208.78 | -207.23 |
| RNA Stability | Decrease | Without change | Decrease |
| RNA Structure Change | + | - | + |
| Expasy Prediction for the Protein Hydrophobicity | |||
| Amino acid Change | Hydrophobic to hydrophilic | Polar to non-polar | Neutral to Hydrophilic |
| Hydrophobicity Chnngea | F: 2.800 to Y: -1.300 | T: -0.700 to A: 1.800 | G: -0.400 To R: -4.500 |
| Expasy prediction | Affected | Affected | Affected |
| SIFT | Tolerated | Tolerated | Deleterious |
| PolyPhen-2 | Probably Damaging | Benign | Probably Damaging |
| I-Mutant2.0 /DDGb | Increase Stability/0.87 | Decrease Stability/-1.54 | Decrease Stability/-2.06 |
| PhD-SNP | Neutral 78% | Neutral 89% | Deleterious 59% |
| PROVEAN | Neutral | Neutral | Deleterious |
| PredictSNP | Neutral 75% | Neutral 83% | Deleterious 52% |
| ClusPro 2.0, (Effects on Protein Docking) | + | - | + |
Prediction of Missense Mutations as Effective Variations by the Functional Prediction Tools.
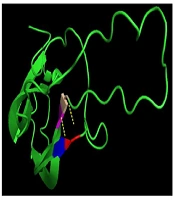
In the present study, a three-dimensional structure model was used to predict the TSHβ protein using PyMol (Python-enhanced molecular graphics tool), and the results indicated that p.G106R mutation is part of a coil structure located in a conserved region among several mammalian species (Figure 4).
. A, The results of this model show that glycine connects with valine and cysteine with two bonds. B, In the mutant protein, arginine binds to the two amino acids of cysteine and one valine with three bonds. C, The position of p.G106R mutation in TSHβ protein indicated that this mutation is part of a coil structure.")
The prediction of the tertiary structure of TSHβ protein by PYMOLE for the normal and mutant variant (p.G106R mutation). A, The results of this model show that glycine connects with valine and cysteine with two bonds. B, In the mutant protein, arginine binds to the two amino acids of cysteine and one valine with three bonds. C, The position of p.G106R mutation in TSHβ protein indicated that this mutation is part of a coil structure.
5. Discussion
CCH is a rare kind of congenital hypothyroidism with genetic causes, mostly in the TSHβ subunit gene. TSH is a 28 kDa glycoprotein hormone released by the anterior pituitary gland. Similar to other glycoprotein hormones, TSH is a heterotrimeric protein consisting of a common α-subunit (92 aa) and a specific β-subunit (118 aa). The TSHβ gene with 4.9 kb in size contains 3 exons and 2 introns. The first exon contains 37 bp and includes the 5' untranslated region of the gene. The coding region of the gene includes the second exon (163 bp) and the third exon (326 bp), which are separated by a 450 bp intron (15).
For the TSHβ gene, more than ten different mutations have been identified so far, which all are considered as probable causes of TSH deficiency worldwide. Inheritance of CCH is autosomal recessive (17). Because in most screening programs, attention is only paid to high TSH levels, and fT4 is often ignored, the diagnosis rate of CCH is often low. Therefore, diagnosis and initiating thyroid hormone replacement may be delayed in these children, resulting in neurodevelopmental retardation and growth failure (18). Like other CCH pediatric patients with late diagnosis, our patients were also diagnosed between the ages of 3.0 - 6.5 years (Mean ± SD = 4.42).
In the present paper, using a PCR-based direct sequencing strategy, we described three different missense mutations in the TSHβ -subunit gene in five (out of seven) Iranian unrelated children patients affected by congenital TSH-deficient hypothyroidism. Two heterozygous missense mutations (c.32T>A: p. F11Y and c.316G>C: p. G106R) are novel and so far has not been detected in any patients with low TSH hypothyroidism. Also, three affected children were homozygous for A-to-G substitution in codon 40 of the TSHβ subunit that leads to the conversion of threonine to an alanine (p.T14A), a common missense variant in the TSHβ gene.
The F11Y mutation in the conserved SMLFGL region of exon 2 of the gene was identified in a six-year-old boy. This new mutation constitutes a T to A transversion (c.32T>A) and results in a change from nonpolar phenylalanine to polar tyrosine in the mutant peptide. The heterozygous (F11Y) and homozygous (T14A) missense mutations found in exon 2 of the TSHß gene, causing amino acid substitutions in the signal peptide of the TSHß-subunit. The observation of these mutations possibly indicates the presence of new mutational hot spots of the TSH ß gene. TSHß-subunit-like other secretory proteins are synthesized as precursors extended at the NH2 terminus by a sequence of 20 amino acids, termed the leader or signal sequence, which is required for translation by ribosomes and protein transport from the membrane. The sequences of signal peptides are heterogeneous, but mostly, the signal peptides have consisted of three conserved domains: a hydrophilic NH2 terminal region of five to eight positively charged basic amino acids, which is essential for translocation, a central hydrophobic core of 7 to 15 amino acids, which is vital for recognition and transferring precursor protein to the ER membrane, and a polar COOH terminal region of approximately six amino acids, which affects the fidelity and efficiency of signal peptidase cleavage as posttranslational processing (19).
In the present study, the c.32T>A mutation of the TSHß gene was located in the hydrophobic core of the signal peptide, causing an F11Y amino acid substitution. According to the in-silico prediction results, this missense mutation changes the hydropathy determinant of the core region (hydropathy score alters from 2.800 for nonpolar and hydrophobic phenylalanine to -1.300 for polar and hydrophilic tyrosine). Thus, it probably leads to impairment of the recognition and transformation of precursor protein into the endoplasmic reticulum. Also, amino acid sequence alterations near the cleavage site in the COOH terminal region of the signal peptide can disrupt the efficiency of signal peptidase cleavage. The c.40A>G missense variation (T14A) is located in the polar COOH terminal region and changes the hydropathy determinant of this region (hydropathy score alters from -0.700 for polar threonine to 1.800 for non-polar alanine), so it can influence the hydrophobic interactions.
Furthermore, the G106R mutation in exon 3 of the gene was identified in a 3.5-year-old affected girl. In previous studies, exactly a nucleotide before G106R mutation, C105V mutation has been identified in related and unrelated cases from different populations such as Brazilian (20), German (21, 22), and Belgium (23) patients. Due to the high frequency of this mutation in different populations, it’s argued that this region is critical for maintaining the biological and structural integrity of the TSH molecule and represents a mutational hot spot (11, 24). Furthermore, the G106R mutation changes the amino acid sequence in the seat belt region of the TSH heterodimer, the important region for the dimerization with the ɑ-subunit and essential for the accurate secretion of the mature TSH. In-silico analysis for novel mutation c. 316G>C revealed the pathogenic nature of this alteration (PolyPhen2: 0.568, the score of SIFT: 0, and Mutation Taster prediction: disease-causing). The prediction model of the tertiary structure of TSHβ protein also indicated that this mutation is part of a coil structure in the protein. This model shows that in the position of 106, glycine amino acid (a small and without side-chain amino acid) connects with valine and cysteine at positions of 71, and 72, respectively. These bonds are conserved and located in important positions of this protein. In the mutant protein, the amino acid arginine has a side chain with three carbons and changes the number of bands in this position, as it binds to the two amino acids of cysteine and valine at positions 105, 72, and 71, respectively. The findings of the prediction model of the tertiary structure of mutant TSHβ protein suggest that the conformation of the TSHβ subunit is probably altered with this mutation.
The other confirmatory evidence that this mutation is a pathogen cause of CCH in our patient roots in the fact that this mutation was not identified in 60 ethnically matched healthy subjects. The interpretation of measurable TSH in parents of this case was difficult. Particularly because both parents did not show any mutation in this region. Nevertheless, early clinical data of this patient with poor growth indicated low serum FT4 and TSH hypothyroidism as well as normal basal values of cortisol and prolactin. Besides, no response to TRH was observed, which in total revealed the necessity of molecular analysis. However, in all affected patients, central congenital hypothyroidism was not distinguished by neonatal screening when only the TSH was considered so that the exact diagnosis of CCH due to TSH deficiency was delayed. Because severe mental retardation has been reported in some children with CCH, using early molecular diagnosis in-time thyroid hormone replacement therapy can be provided.
The current study had limitations, including the necessity of evaluating the frequency of the mutations in a study with a larger sample size. Secondly, inaccessibility of information regarding the family history of the CCH for all participants. Moreover, applied studies are needed to investigate the contribution of these novel variants to the CCH phenotype.
In conclusion, three missense mutations (two novels and one previous reported) in exons 2 and 3 of the TSH ß-subunit gene in 5 sporadic cases of CCH are reported. The findings of the prediction model demonstrated that these mutations may affect the structure of TSH, resulting in impairments in the biological functions of the protein. According to the best knowledge of the authors, this study is the first of its kind in Iran, and our observations suggested that these variations may be associated with non-familial CCHs phenotypes in Iranian pediatric patients. Nevertheless, since the sample size was small, such an association cannot be confirmed. Also, the findings showed that these missense mutations may be more common than previously thought in CCH patients. However, additional studies are required to identify the mechanisms by which these novel mutations lead to genomic instability and contribute to the risk of sporadic CCH in the Iranian population.
