



1. Background
Bovine herpesvirus-1 (BHV-1) is a member of Herpesviridae (1) with a 135301 bp genomic dsDNA. In the genome, there are four segments including a unique long (UL) segment, a unique short (US) segment, and two inverted repeat regions, namely inverted internal repeat (IR) and terminal repeat (TR) sequences. The genome encodes for 70 proteins. Thirty-three proteins are structural, 13 of which are present in the viral envelope and 10 are capable to encode viral glycoproteins. Six genes that encode for six different glycoproteins are located in the UL segment (2). The UL25 gene is located at 60602 - 62398 position. Its product is a tegument protein, which plays an important role in the assembly and packaging of the genome into the capsid (3).
Bovine herpesvirus-1 is classified into three subgroups including BHV-1.1, BHV-1.2 and BHV-1.3. BHV-1.1 is responsible for the respiratory manifestations of the disease (IBR) (4, 5).
Over the years, a great deal of research to develop anti-IBR vaccines has led to the fact that several vaccines are now available for this disease. However, none of them are completely effective. Therefore, the development of specific and adequate methods for the prevention and treatment of BHV-1 is great of necessity (6).
Since conventional therapies for the treatment of human and animal viral diseases have their limitations and alternative therapies are urgently needed, gene therapy techniques, such as RNAi technology have been used as potential therapeutic approaches. One way to evaluate the efficacy of gene therapy is to provide reporter cell lines for the expression of viral target genes. Therefore, the first step is to clone the viral target sequences into a suitable vector for transfer to the appropriate cell line.
In the previous studies, due to the conservation of the BHV-UL25 and its functional roles, it has been frequently selected as a target. Thus, in the present study, the conserved BHV-UL25 serine protease substrate domain was selected for cloning in a lentiviral plasmid (7).
Over the recent years, gene therapy via lentiviral vectors (LV) has increased dramatically. Lentiviral vectors based on the human immunodeficiency virus1 (HIV-1) are intended as a vector with high capabilities for gene transfer studies. They can integrate and transfer genes into non-dividing cells. The pseudotyped envelope with VSV-G protein increases the types of host cells. To create such a vector, the most common method is to clone the essential virion and transgene genes into different plasmids and consequently, the co-transfection of them to the appropriate cell line. Accordingly, the possibility of generating replication-competent lentivirus will be very low (8, 9).
Lentiviruses can induce their target gene expression for a long time. In addition, they are able to transfer large nucleotide sequences. Furthermore, because of the low probability of integration into or next to the cell’s oncogene, the possibility of mutagenesis and carcinogenesis following gene induction by lentiviral vectors is lower than that by other vectors (10).
Due to the mentioned reasons mentioned above, in the present study, lentiviral vectors were preferred for the cloning of the desired gene in a lentiviral transfer vector.
Among the available lentiviral plasmids, pCDH-CMV-MCS-EF1-cGFP-T2A-Puro, which has ampicillin and puromycin resistance genes, was selected for ease of access and handling. Both antibiotics are easy to achieve. Moreover, the vector has a GFP gene as a marker protein that is applicable for the evaluation of transfection efficiency. In addition, this plasmid passes strong RSV, CMV, and EF1 promoters for expression of the desired gene.
2. Objectives
The objective of this study was to produce a pCDH-CMV-MCS-EF1-cGFP-T2A-Puro lentiviral plasmid expressing BHV-UL25 serine protease substrate domain as a transfer vector.
3. Methods
3.1. Preparation of Serine Protease Substrate Fragment of BHV1-UL25
The sequence of BHV1-UL25 serine protease substrate domain was obtained from National Center of Biotechnology Information (NCBI) database (Ac. No. AJ004801.1) and aligned with all the known gene sequences in other BHV1 strains extracted from NCBI. The alignment was performed using the online software www.ebi.ac.uk/Tools/msa/clusalo.
Afterwards, EcoRI and BamHI cleavage sites were added to the 5’ and 3’ ends, respectively. The His Tag sequence was added to 3’ end for future monitoring tests. Finally, this sequence was sent to the Shine Gene Molecular Biotechnology Company (China) for synthesis and clone in pGH pBLUSCRIP (+) vector.
3.2. Restriction Enzyme Digestion
pCDH-CMV-MCS-EF1-cGFP-T2A-Puro lentiviral plasmid (Addgene) and pGH pBLUSCRIP (+) carried BHV1-UL25 serine protease substrate fragment digested by BamHI (Roche, Germany, Cat. No: 10220612001) and EcoRI (Roche, Germany, Cat. No.: 10220566001) for extraction of the inserted fragment. Therefore, 7.5 μL of plasmid (2 μg/μL), 1.3 μL of each enzyme (10 U), 2.5 μL of 10X K buffer, and nuclease-free water were added to a final volume of 25 μL. After incubation at 37°C for 1 hour, 1 μL of digested plasmid was electrophoresed in a 1% agarose gel in order to confirm the restriction enzyme digestion.
3.3. Extraction of BHV1-UL25 Serine Protease Substrate Fragment from Agarose Gel
In order to purify the digested plasmid, after digestion with EcoRI and BamHI and removing the undigested plasmids and restriction enzymes, the product of digestion was extracted from agarose gel using Expin Gel SV (50 prep) kit (Gene all, Cat. No.: 000001422) according to the manufacturer’s instructions.
3.4. Ligation of BHV1-UL25 Serine Protease Substrate Fragment into Lentiviral Plasmid and Transformation
For the ligation of the inserted fragment into vector 2 μL of T4 10X ligation buffer (Fermentas, Germany, Cat. No.: B69), 2 μL of 50% PEG-4000 (Fermentas, Germany), 1 μL of T4 DNA ligase (Fermentas, Germany, Cat. No.: EL0013), 150 ng of the inserted DNA, and 50 ng of plasmid were mixed with nuclease-free water up to 20 μL total volume. Heat shock protocol was applied for the transformation of ligation mixture. In summary, 10 μL of ligation product was added to 100 μL of DH5α. Subsequently, the mixture was placed in the following temperature conditions: 30 min at 4°C, 90 s at 42°C, and 5 min at 4°C. Afterwards, 900 μL of LB broth was added and the mixture was shaken at 200 rpm for 45 min and centrifuged at 6000 rpm for 3 min. Finally, the culture of pellet in LB agar +100 mg mL-1 ampicillin was performed and put at 37°C for 14 - 16 hours.
3.5. Colony PCR
After culturing the transformed bacteria, PCR reaction and mini preparation method of plasmid extraction was performed for 10 individual clones of each plate. PCR was carried out with the general primers of pCDH (CMV-F: AATGGGCGGTAGGCGTGTA-3’and EF1-R: 5’-GGACTGTGGGCGATGTG-3’). The thermal cycling conditions was as follow: 95°C for 4 min, 30 cycles at 95°C for 30 s, 55°C for 35 s, 72°C for 45 s, and one cycle at 72°C for 9 min. The positive BHV1-UL25 serine protease substrate fragment that was synthetized and cloned in pUC57 vector by Shine Gene Molecular Biotechnology Company (China) and negative controls (deionized water) were employed in each test. The PCR reaction was included of 12 μL of dW, 9 μL of Taq DNA Polymerase Master Mix Red 2x -2 mM MgCl2, (Amplicon, Cat. no. A180301), 1 pmol of each primer, and 2 μL of DNA template up to 25 μL total volume.
3.6. Sequencing
After electrophoresis in 0.8% agarose gel, two reactions of colony PCR‑positive samples, which contained the recombinant plasmids, were sent to Bioneer Company (Korea) for sequencing.
4. Results
4.1. Extraction of BHV1-UL25 Serine Protease Substrate Fragment from Agarose Gel
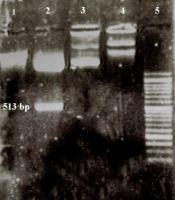
One μL of digested plasmid and 1 μL of the uncut plasmid were electrophoresed in a 1% gel along with an Excel Band™ 1 KB Plus (0.1 - 10 kb) DNA ladder and the extraction of the fragment from digested pGH pBLUSCRIP (+) and the difference of size between cut and uncut plasmids were observed (Figure 1).
 and pCDH-CMV-MCS-EF1-cGFP-T2A-Puro; (1) Digested pCDH (8197 bp); (2) Digested pGH pBLUSCRIP II SK-UL25 (+); the band above: Digested plasmid (2943 bp), bottom band: BHV1-UL25 serine protease substrate fragment (513 bp); (3) Uncut pCDH (8220 bp); (4) Uncut pGH pBLUSCRIP II SK-UL25 (3456 bp); (5) 50 bp DNA ladder")
Extraction of digested and uncut pGH pBLUSCRIP II SK-UL25 (+) and pCDH-CMV-MCS-EF1-cGFP-T2A-Puro; (1) Digested pCDH (8197 bp); (2) Digested pGH pBLUSCRIP II SK-UL25 (+); the band above: Digested plasmid (2943 bp), bottom band: BHV1-UL25 serine protease substrate fragment (513 bp); (3) Uncut pCDH (8220 bp); (4) Uncut pGH pBLUSCRIP II SK-UL25 (3456 bp); (5) 50 bp DNA ladder
4.2. Colony PCR
After colony PCR, the presence of the BHV1-UL25 fragment was detected in transformed colonies samples with the size of 726 bp (Figure 2). The length of the PCR product in origin pCDH (positive control) was in the range of 213 bp. Moreover, no band was detected in untransformed DH5α (negative control).
 Ladder (50 bp); (2) and (3) PCR products (726 bp); (4) Positive control (213 bp)")
Colony PCR for detection of BHV1-UL25 serine protease substrate; (1) Ladder (50 bp); (2) and (3) PCR products (726 bp); (4) Positive control (213 bp)
4.3. Extraction of Recombinant Plasmids Containing of BHV1-UL25-Serine Protease Substrate
One μL of extracted plasmids from two positive clones were electrophoresed on 1% agarose gel and the production of recombinant vector was confirmed by observing the desired band at 8602 bp (Figure 3).
 - (3) Recombinant pCDH-CMV-MCS-EF1-cGFP-T2A-Puro plasmids in different concentrations (8520 bp); (4) 1 kb ladder")
Extraction of BHV1-UL25 serine protease substrate-pCDH-CMV-MCS-EF1-cGFP-T2A-Puro; (1) - (3) Recombinant pCDH-CMV-MCS-EF1-cGFP-T2A-Puro plasmids in different concentrations (8520 bp); (4) 1 kb ladder
4.4. Sequencing
Analysis of the read sequences of recombinant plasmids showed the presence of the desired fragment and the accuracy of the cloning process (Appendix in supplementary file).
5. Discussion
On the one hand, the effectiveness of existing anti-IBR vaccines or drugs has not been approved by the majority of scientists (6, 11) and on the other hand, treatment based on suppressing or changing the desired gene expression has minimal side effects compared to other treatment methods. Therefore, in recent decades, gene therapy has been welcomed as an alternative method of treatment (12).
Genetics has been involved in the treatment of herpes viruses in the recent years due to the low specificity and adverse effects of chemical drugs. New studies have been developed on the antiviral approaches of gene therapy against these viruses. Accordingly, designing and developing methods for evaluating such treatments is necessary. Hence, in the present study, the early stages of developing an approach to assessing to genetic treatments against BHV-1 were performed.
One way to evaluate such molecules is to introduce them into cell lines that express the genes they are supposed to suppress. For this purpose, the first step is the cloning of these target sequences into an appropriate plasmid for transfection into a particular cell line (13).
IBRV UL25 is a nuclear gene that is conserved among members of the Herpesviride family. Its necessity for the assembly and addition of the tegument layer to the virion encouraged the authors of the present study to select it. In a study performed by Bowman et al., using the UL-25 gene cloning in pET41 plasmid and its expression in cells, the researchers showed the role of UL25 in the formation of a stable capsid and the proper packaging of the viral genome (2). Non-expression of BHV-1 UL25 occurred in MDBK cells. Several works have been conducted to study the sequence and structure of the UL25 gene since then, all of which have shown this gene is conserved among all subfamilies of Herpesviridae (2).
UL25 is a DNA packaging tegument protein and its main role is considered to be DNA encapsidatoin. The placement of the serine protease substrate domain in UL25 indicates that this is the cleavage site of serine proteases that facilitate DNA encapsidatoin, and its conservation indicates it has an important role for the virus life cycle (14).
In the present study, for permanent expression of the target genes, the transduction was performed by a lentiviral vector. Plasmids were also used for gene expression induction after the transfection although the duration of gene expression was low (15). Lentiviral vectors could infect almost all types of cells and induce remarkable sufficient gene expression in their host cells for a long time. Furthermore, owing to their potent to integrate into the host chromatin, their desire to eliminate all pathogenic genes in the vector, and finally, because there is no interference with the pre-existing antiviral reactions of immune system, lentiviral vectors LVs possess an effective delivery. Moreover, LVs can transmit large nucleotide sequences (3000 bp) and the probability of mutagenesis and carcinogenesis is low after their application.
Given the reasons mentioned above, lentiviral vectors were used to express the target genes in the desired cells and the first step for the preparation of these vectors is the cloning of the desired gene in the transfer vector in lentiviral packaging systems (16, 17).
Among the available lentiviral plasmids, pCDH-CMV-MCS-EF1-cGFP-T2A-Puro, which has ampicillin and puromycin resistance genes, was selected for ease of access and handling. Both antibiotics are easy to achieve. On the other hand, the vector has a GFP gene as a marker protein, which is applicable to the diagnosis of transfection efficiency (18, 19). This plasmid passes strong RSV, CMV, and EF1 promoters for expression of the desired gene and GFP. The multiple cloning sites in this plasmid include the cleavage sites of the most restriction enzymes. Jafarzade et al. firstly increased the efficiency of LVs (pCDH-CMV-MCS-EF1-cGFP-T2A-Puro) and epEGFP-N1 for induction their desired gene in 293T cells (20). Ranjibar et al. amplified the Cop-GFP from the pCDH plasmid for developing a phagemid for production of reporter mammalian-cell (21).
There are some applications of RNAi against the members of Herpesviridae. For example, siRNAs were designed by Jia and Sun for suppression of gamma Herpesvirinae ORF-45 to demonstrate its performance. BHK-21 cells expressed it were developed to evaluate these siRNAs (22).
Wiebusch et al. designed siRNAs for HCMV-UL54 and produced U373 cells expressing UL54 sub-replicons after cloning the target gene segments into appropriate plasmids and transfecting them (23).
Zhe et al. targeted HSV-1-UL39 by their siRNAs. They evaluated siRNAs by designing, cloning, and providing an ICP6 reporting system (24).
Wilkes and Kania suppressed FHV-1-gD by siRNA molecules decrease viral titer (25). Specific evaluation of these two siRNAs was performed with gene cloning and plasmid transfusion into Crandell-Rees cat kidney cells (25).
Narute et al. designed two anti BHV1-UL25 siRNAs. However, in their study, they did not provide a proprietary evaluation reporter system (7).
There are some researches in which lentiviruses mediated RNAi have been applied for other viruses Herpesviridae family; Wang et al. suppressed LMP2A with lentivirus-mediated RNAi in GT38 cells (26). Song et al. used lentivectors for expression of IBRV gD shRNAs and with TCID50 assay showed the reduction in viral titer. They transfected pcDNA3-gD plasmid and recombinant LVs expressing shRNA into 293T cells and RNAi efficacy was confirmed utilizing Western blot (6). Amjadimanesh et al. applied BHV1-UL25 shRNAs and showed that they had considerable antiviral attributes in challenge with virus and MDBK cells expressed UL25 (27).
5.1. Conclusions
The findings revealed that the BHV1-UL25 target fragment was successfully cloned in the desired plasmid and in the future studies; it will be accessible for transfection of the appropriate cell lines using lentiviral packing systems to produce specific reporter cells.
